有価証券報告書-第16期(令和2年1月1日-令和2年12月31日)
事業内容
1.当社の事業概要について
(1) 当社の概要
当社は、元米国アムジェン社(注1)本社副社長で、同社の日本法人であるアムジェン株式会社(現在は武田薬品工業株式会社が全事業を譲受)の創業期から約12年間社長を務めた吉田文紀が、2005年3月に設立した医薬品企業です。
経営理念は「共創・共生」(共に創り、共に生きる)で表され、患者さんを中心として医師、科学者、行政、資本提供者を「共創・共生」の経営理念で結び、アンメット・メディカル・ニーズ(Unmet Medical Needs)(注2)に応えていくことにより、社会的責任及び経営責任を果たすことを事業目的としています。
なお、当社の事業は、医薬品等の研究開発及び製造販売並びにこれらの付随業務の単一セグメントであるため、セグメント別の記載を省略しています。
(注1) バイオ医薬品業界最大手。1980年、米国カリフォルニア州サウザンド・オークスにおいて、AMGen(AppliedMolecular Genetics)として設立。日本においては、1993年5月1日にアムジェン株式会社として業務を開始しました。なお、2008年2月に武田薬品工業株式会社がアムジェン株式会社の株式を100%取得後、現在は武田薬品工業株式会社が全事業を譲受しています。
(注2) アンメット・メディカル・ニーズ(Unmet Medical Needs)とは、未だ満たされない医療上の必要性を意味し、患者さんや医師から強く望まれているにもかかわらず有効な既存薬や治療がない状態を指します。
(2) 当社の事業の特徴
がん及び血液領域における希少疾病分野(注3)の研究開発の多くは、欧米を中心に、大手製薬企業よりもむしろ、多くの大学・研究所、バイオベンチャー企業により創薬研究・新薬開発が活発に行われ、海外では既に数々の有用な新薬が医療の現場に提供されています。しかし、これらの分野は開発に高度の専門性が求められることから、開発の難度も高く、また大手の製薬企業が事業効率の面、採算面で着手しにくいため日本を初めとするアジア諸国においては手掛けられていない空白の治療領域となっています。当社は、極めて医療上のニーズは高いものの、新薬の開発が遅れている空白の治療領域をビジネスチャンスと捉え、特に、高い専門性が求められ難度が高いために参入障壁の高いがん及び血液領域を中心とした日本初のスペシャリティ・ファーマ(注4)です。当社は、大型新薬(いわゆる売上高が1,000億円を超える「ブロックバスター」)の追求ではなく、マーケットは相対的に小規模でも医療ニーズの高い希少疾病分野を中心とした新薬開発に取り組み、これらの医薬品及び新薬候補品を数多く保有することにより、強固なパイプライン・ポートフォリオを構築し、高付加価値で高収益を達成し、持続性のある事業展開を行います。
当社は、このような空白の治療領域を埋めるための新薬の開発・提供を行うことを企業使命として設立されました。新薬が開発されないことで治療上の問題を抱えている患者さんに対して、短期間で開発をし、迅速に治療薬をお届けすることを最優先に考え、医療への貢献、そして医薬品業界の健全な発展に寄与することにより、持続的成長と安定への道を進んでまいります。
(注3) 希少疾病分野とは、患者数が少ない疾病分野のことで、この分野に対する医薬品は希少疾病用医薬品(Orphan Drug:オーファンドラッグ)と呼ばれます。厚生労働省はオーファンドラッグ制度を設定し、我が国において患者数が5万人未満の重篤な疾病であること、医療上特にその必要性が高いこと等をその指定の基準としています。当該指定を受けると、申請から承認までの期間が短縮され、再審査期間が最長10年になる等の優遇措置があります。
(注4) スペシャリティ・ファーマとは、得意分野において国際的にも一定の評価を得る研究開発力を有する新薬開発企業をいいます(2007年「新医薬品産業ビジョン」(厚生労働省)の定義による)。
(3) 当社の事業モデルについて
創薬系事業の特徴として、新薬の開発は長期間にわたり膨大な先行投資を強いられるものの、その研究開発の成功確率は極めて低いことが知られています。一般に、研究所において何らかの生物・生理活性(注5)が認められた化合物が新薬として承認にいたる確率は、2万分の1~2万5千分の1と言われています。また、承認を取得した新薬のうち、上市・販売後において採算が取れるのはそのうちの15~20%以下と言われています。当社は、このような創薬系事業の難しさを踏まえた事業モデルを構築しています。
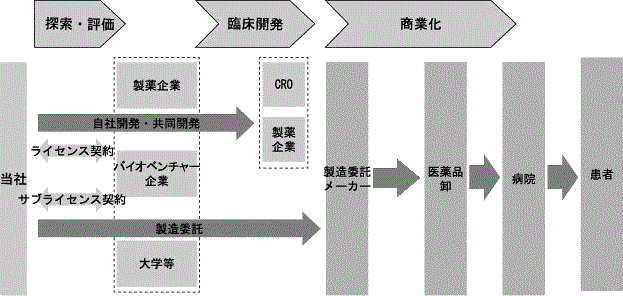
当社では、開発にかかる様々なリスクと費用を軽減するとともに、開発候補品の臨床試験を迅速・確実に進め、開始から承認取得までの期間を短縮するために、主として既にヒトでPOC(Proof Of Concept)(注6)が確立され、前臨床試験データと臨床試験データがある化合物を対象としております。これらの化合物の探索は当社独自の探索ネットワークと評価ノウハウを活用して、社内の経験を有した専門スタッフによる第1次スクリーニングにより絞り込みを最初に行います。その後、科学的諮問委員会(Scientific Advisory Board:以下「SAB」といいます)(注7)において、第一線で関連分野における治療の研究に携わる経験豊かな社外専門家の厳密な評価を受けた上で、当社において最終的な導入候補品を決定いたします。
社内外の専門家による、こうした“目利き”のプロセスを経て、当社はがん及び血液領域を中心として、製薬企業、バイオベンチャー企業等から主にヒトでPOCが確立された開発品の日本並びにアジア諸国、さらには欧米を含むグローバルの開発・製造・販売権を継続的に確保することにより、持続性のある事業を展開しています。そのような、開発の成功確率が高く、事業性のある、魅力的な開発候補品を導入するためには、この“目利き”の力に加え、がん及び血液という開発の難度が高い治療領域における当社の開発力について、開発候補品の提供者であるライセンサーから高い評価を得ることも導入の成否を決める重要なポイントとなります。そのためには、①適切な治験計画の策定、②治療対象となる適切な治験患者の選定、③その領域における医学専門家と公正な関係を維持・構築できる、専門性の高い優秀な開発スタッフが必要となります。これらの総和が開発力となり、開発を着実に、かつ迅速に実行することが可能となります。がん及び血液分野で実績のある大手製薬企業の開発部門で経験を積んだ人材を中心に構築された当社の開発チームが導入から承認申請までを僅か4年間という短期間でなし得た、抗がん剤 SyB L-0501での実績は、ライセンサー、パートナー企業、導入候補先企業から高い評価を得ています。
なお、開発につきましては、基本的な開発戦略の中枢となる臨床試験のデザイン、海外の試験との連携、医学専門家との調整等は当社が主体となって手掛け、定型的な開発業務は、外部資源であるCRO(Contract Research Organization 受託臨床試験実施機関)(注8)へ業務委託し、製造についてはライセンス供給元あるいは信頼できる国内外の製薬企業へ業務委託を行います。
販売につきましては、2008年8月に締結した事業提携契約に基づき、エーザイ株式会社(以下「エーザイ」という)を通じて国内販売を行ってまいりました。事業提携契約が2020年12月に満了となることから、2018年10月よりトレアキシン®の国内販売について自社による販売体制構築の準備を開始しました。2020年12月からの自社販売体制への移行に向けて、がん及び血液領域に精通した自社MR(Medical Representative)(注9)を中核とした全国営業体制の構築と流通及び物流機能の整備を推進すると同時に営業戦略・企画の策定及び市場調査を行うマーケティング体制の強化に努めるとともに関係治療領域におけるKOL(Key Opinion Leader)(注10)との良好な関係構築、的確な医療ニーズの把握と市場調査を行い、各種データ、ノウハウの蓄積を図ってまいり、2020年12月の契約満了に伴い自社販売体制へ移行しました。
これらの事業モデルを図示すると以下のようになります。
(注5) 生理活性とは、化学物質が生体の特定の生理的調節機能に対して作用する性質のことです。この生理活性の作用を持つ化学物質を疾病治療に応用したものが医薬品となります。
(注6) POC(Proof of Concept)とは、新薬候補物質の有効性や安全性を臨床で確認し、そのコンセプトの妥当性を検証することを意味します。
(注7) 科学的諮問委員会(SAB:Scientific Advisory Board)とは、世界中から集まる膨大な新薬候補を元に、医療ニーズの高さや収益性などリスクバランスのとれたポートフォリオを、それぞれの専門の立場から意見や提言を交え徹底的に議論した上で、パイプライン戦略を構築する、当社の重要な評価機関です。当社では、SABを年2~3回開催し、世界中から優れた実績と経験をもつ臨床医・基礎科学者の方々に、当社の創薬研究及び新薬開発のアドバイザーとして参画いただいています。
(注8) CRO(Contract Research Organization)とは、製薬企業が、自社で実施する開発業務を遅滞なく進めるために、一部の業務について委託を行う機関です。委託業務の内容としては、治験が実施計画書どおりに遂行されているかをモニタリングするモニター業務や、臨床データを管理するデータ管理業務などがあります。
(注9) MR(Medical Representative)とは、自社医薬品に関する情報の専門家として医療機関を訪問し、医療関係者と面談することにより、医薬品の品質・有効性・安全性等に関する情報の提供・収集・伝達を主な業務とする医療情報担当者をいいます。
(注10)KOL (Key Opinion Leader)とは、担当領域の治療において他の医師に影響力を持つ医師のことをいいます。
(4) 当社の事業戦略
当社は、上記の事業を成功させるために、主に以下の5つの事業戦略を展開しています。
(a) ポストPOC戦略による開発リスクの軽
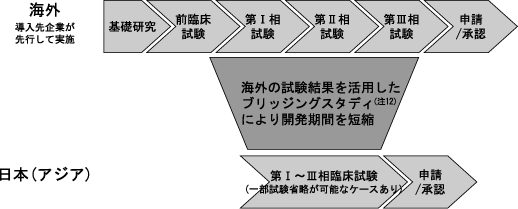
当社の導入候補品(注11)は、主として既にヒトでPOCが確認されていることを原則としています。従って、臨床開発ステージが比較的後期段階にある候補品か、既に海外で上市されている製品が対象となります。これらの導入候補品は既に海外で先行して開発が行われており、新薬としてヒトでの有効性・安全性が確認されていることから、開発リスクを軽減でき、また、先行している海外の治験データを活用することにより日本を含めアジアにおける開発期間を短縮するとともに開発コストを低減し、成功確率を高めることが可能となります。
(注11)導入候補品とは、当社の開発候補品として他社より開発権等の権利取得を検討している化合物を指します。
(注12)ブリッジングスタディとは、外国での臨床データを活用するために国内で行われる試験のことをいいます。この国内試験の結果を外国のデータと比較し、同様の傾向があることを確認します。
(b) 高度な探索及び評価能力による、優れたパイプラインの構築
当社の新薬サーチエンジンは、製薬企業及びバイオベンチャー企業等との多様なネットワークによって構築され、膨大な化合物の中から、社内の専門家による厳正な評価を経て、有望な導入候補品が抽出されます。これらの導入候補品は更に、第一線で研究に携わる経験豊かな専門家により構成されるSABに諮られ、そのアドバイスと評価を受けた上で導入候補品を決定しています。この開発品導入決定までの高度なスクリーニングプロセスは、既に海外において有効性・安全性が確認された開発品を導入するポストPOC戦略と相まって開発リスクの軽減と開発期間の短縮につながることになり、また、候補品が医療の現場において求められるものかどうかの医療ニーズの充足度に対する理解、及び上市後の収益予測の精度向上に貢献しています。
<当社の開発品導入プロセス>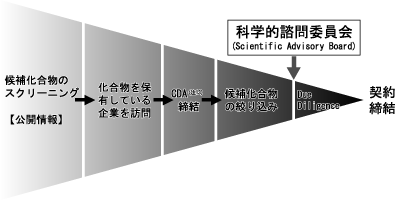
(注13)CDAとは、Confidential Disclosure Agreementの略で、秘密保持契約書のことを意味します。
(c) ラボレス・ファブレス戦略による固定費抑制
当社は、一切の研究設備や生産設備を保有していません。研究設備・生産設備はともに固定費発生源の代表格ですが、当社はこれらを一切保有せずに、開発候補品の探索及び導入後は、開発品の開発戦略策定と実行等の付加価値の高い業務に専念し、そのほかに必要とされる定型的な開発業務は外注しています。これにより低コストの医薬品開発を実現するとともに、財務戦略の機動性を確保しています。
(d) ブルーオーシャン戦略(注14)による高い事業効率の実現
海外で標準治療薬として使用されている製品が日本では使用できない、あるいは海外で新薬として承認された製品が5年近くも遅れて日本で承認される、いわゆるドラッグ・ラグの問題が深刻化しており、がん患者の難民という言葉も生まれています。このドラッグ・ラグは、当社の戦略的開発領域である難治性のがん及び血液疾患領域で特に目立っています。特に抗がん剤の市場自体は大きく、また高齢化に伴い現在も拡大傾向にあるものの、抗がん剤の対象疾患は多岐にわたり、がん腫により細分化されているため、各々のがん腫でみると対象患者数がそう多くはない治療領域が数多く存在します。これらの領域での新薬の開発には、極めて高い専門性が求められ、開発の難度が高い半面、大手の製薬企業では採算性などの問題から開発に着手しにくいことがその理由のひとつといわれています。しかし、ひとたび、そうした領域において新薬の承認を取得し上市できれば、競合が少ないため、これらの領域で適応拡大・新製品上市を着実に積み上げていくことで、高成長・高収益を実現できるものと考えています。
(注14)ブルーオーシャン戦略とは、競合との熾烈な競争により限られたパイを奪い合う市場(レッドオーシャン)を避け、市場を再定義し、競合のいない未開拓な市場(ブルーオーシャン)を創造することで、顧客に高付加価値を与えつつ利潤の最大化を目指す戦略です。
(e) アジアからグローバル展開へ
当社はこれまで日本を中心としたアジア各国を対象に事業を展開してまいりました。しかしながら、日本の医療を取り巻く環境が大きく変わっていくなか、アジアに留まっていては大きな発展は望めません。今後はグローバルな展開を視野に入れた開発候補品の探索及び評価を実施してまいります。2019年9月30日にはキメリックス・インク社(本社:米国ノースカロライナ州、以下「キメリックス社」という)との間で抗ウイルス感染症治療薬ブリンシドフォビルに関しての独占的グローバルライセンス契約を締結し、当社は天然痘疾患を除くすべての疾患を対象とした世界全域における開発・販売に加えて製造を含む独占的権利を取得しました。
当社は、従来の日本への候補品の導入に限定せず、グローバルな市場への展開を目指して、開発に着手しております。抗ウイルス感染症治療薬ブリンシドフォビルにつき、播種性アデノウイルス(AdV)感染症及び免疫不全状態でのアデノウイルス(AdV)感染症を対象に、2021年3月10日、米国及び英国を中心とした国際共同第Ⅱ相臨床試験(フェーズ2a)を開始するため、米国についてFDAにINDの申請を行いました。
2.当社のパイプラインについて
当社は現在開発中のパイプラインとして、SyB L-0501、SyB L-1101、SyB C-1101、SyB L-1701及びSyB L-1702、SyB V-1901を有しています。今後も開発候補品を継続的に導入することにより、パイプラインのより一層の拡充及びリスク・リターンのバランスのとれたパイプライン・ポートフォリオを構築してまいります。
<当社パイプラインの進捗状況>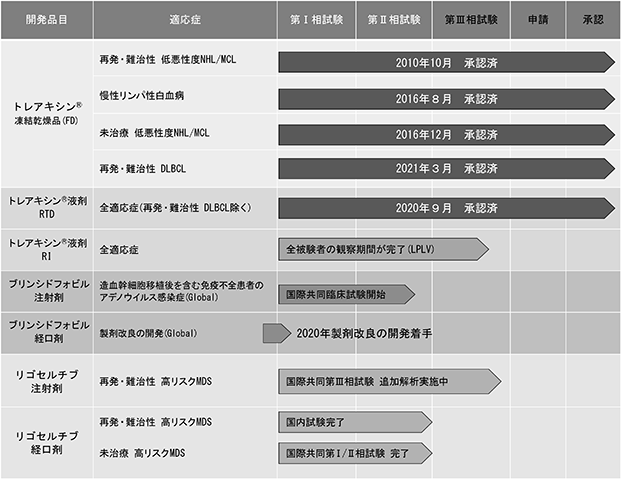
(1) [抗がん剤 SyB L-0501(凍結乾燥注射剤) / SyB L-1701(RTD製剤) / SyB L-1702(RI製剤))(一般名:ベンダムスチン塩酸塩またはベンダムスチン塩酸塩水和物、製品名:トレアキシン®)]
SyB L-0501の主成分であるベンダムスチン塩酸塩(一般名)は、ドイツにおいて非ホジキンリンパ腫(注15)、多発性骨髄腫及び慢性リンパ性白血病の治療薬(商品名「リボムスチン®」)として長年使用されている抗がん剤です。この製品の導入の背景としては、再発・難治性の低悪性度非ホジキンリンパ腫及びマントル細胞リンパ腫の患者さんには、この分野には優れた薬剤がなく、まさしく当社の企業使命である、空白の治療領域を対象とした薬剤であること、また当社の強みである分野(血液がん)であることが導入の決め手となりました。
2018年7月には日本血液学会が発行した造血器腫瘍診療ガイドラインにトレアキシン®とリツキシマブの併用療法(BR療法)が新たに収載され、既承認(再発・難治性の非ホジキンリンパ腫(低悪性度NHL)及びマントル細胞リンパ腫(MCL)、未治療(初回治療)の低悪性度NHL及びMCL並びに慢性リンパ性白血病(CLL))のすべての適応症において、標準的治療の選択肢として推奨されることになりました。これにより名実ともにトレアキシン®が悪性リンパ腫における標準療法として位置づけられています。
既に承認を取得した適応症に続き、再発・難治性のびまん性大細胞型B細胞リンパ腫(r/r DLBCL)を対象とするBR療法による第Ⅲ相臨床試験については、試験成績の主要評価項目である奏効率において期待奏効率を上回る良好な結果が得られたことを基に、2020年5月に製造販売承認事項一部変更承認申請し、2021年3月に承認を取得しました。
2017年9月にイーグル社との間で日本における独占的ライセンス契約を締結したトレアキシン®液剤(RTD製剤及びRI製剤(注16))については、RTD製剤は2020年9月18日に製造販売承認を取得し、2021年1月より販売を開始しました。RI製剤につきましては現在、安全性に関する臨床試験を実施中で、今年度中に承認申請の予定です。RTD製剤は、従来の凍結乾燥注射剤に比べて、手動による煩雑な溶解作業が不要で、そのために要する時間を短縮することができ、医療従事者の負担を大幅に低減することが可能となります。また、RI製剤は、投与時間が、従来の凍結乾燥注射剤及びRTD製剤の60分に対して10分間と大幅に短縮されるため患者さんと医療従事者の負担を大幅に低減することが可能となることから大きな付加価値を提供することができます。更には、液剤の製剤ライセンスによる複数の特許保護を通じてトレアキシン®の製品寿命を2031年まで延長し、当社事業の成長基盤をより強固なものとすることが可能となります。
(注15) 非ホジキンリンパ腫とは、白血球の中のリンパ球ががん化した悪性腫瘍である悪性リンパ腫のうち、ホジキンリンパ腫以外の総称です。日本人の悪性リンパ腫では、大半を非ホジキンリンパ腫が占めています。
(注16) RTD製剤及びRI製剤は、従来の凍結乾燥注射剤(FD)とは異なり既に液化された製剤です。RTD製剤(Ready To Dilute)は調剤作業を大幅に低減し、さらに急速静注であるRI製剤(Rapid Infusion)により点滴時間を従来の60分間から10分間に短縮することにより、FD製剤に比べ患者さんの負担を大幅に軽減し、さらには医療従事者に大きな付加価値を提供することが可能になります。
(2) [抗がん剤 SyB L-1101(注射剤) / SyB C-1101(経口剤)(一般名:Rigosertib Sodium<リゴセルチブナトリウム>)]
リゴセルチブ注射剤については、導入元であるオンコノバ・セラピューティクス社(本社:米国ペンシルベニア州、以下「オンコノバ社」という)が、現在の標準治療である低メチル化剤による治療において効果が得られない、治療後に再発した、または低メチル化剤に不耐容性を示した高リスク骨髄異形成症候群(高リスクMDS)における全生存期間を主要評価項目として、国際共同第Ⅲ相臨床試験(INSPIRE試験)を実施しておりますが、2020年8月に医師選択療法との比較において主要評価項目を達成しなかったことを発表しました。当社は日本における臨床開発を担当しており、INSPIRE試験の追加解析から得られた知見を今後のリゴセルチブの開発に活用するための検討を進めてまいります。
トレアキシン及びリゴセルチブに関して、東京大学医科学研究所との共同研究等を通じて、両化合物あるいは他の既存薬との併用により新たな有用性を見出すとともに新規適応症の探索を行い、事業価値の最大化に努めます。
(3) [抗ウイルス薬 SyB V-1901(一般名:Brincidofovir<ブリンシドフォビル>)]
当社は2019年9月30日にキメリックス社との間で抗ウイルス薬ブリンシドフォビルの注射剤及び経口剤(SyB V-1901、以下各々「BCV IV」及び「BCV Oral」という)(注17)に関しての独占的グローバルライセンス契約を締結し、天然痘疾患を除くすべての疾患を対象としたBCVの世界全域における開発・販売に加えて製造を含む独占的権利をキメリックス社から取得しております。当社は、2020年2月に開催したグローバルアドバイザリーボードでの検討の結果、「空白の治療領域」でアンメット・メディカル・ニーズの高い造血幹細胞移植後のアデノウイルス(AdV)感染症を対象に、日本/アメリカ/ヨーロッパを中心としたBCV IVのグローバル開発を優先的に進めることを決定しております。そして、当該試験により得られた有効性と安全性に関する知見に基づき、造血幹細胞移植後の各種dsDNAウイルス(注18)感染症に対する効果を検討し、抗マルチウイルス感染症へ対象領域を拡大し、更には腎臓移植を含む臓器移植分野等の対象領域拡大の可能性を追求することで、市場の拡大とBCVの事業価値の最大化を目指してまいります。BCV液剤の小児のアデノウイルス感染症を対象とした試験開始に向けて、播種性アデノウイルス(AdV)感染症及び免疫不全状態でのアデノウイルス(AdV)感染症を対象に、2021年3月10日、米国及び英国を中心とした国際共同第Ⅱ相臨床試験(フェーズ2a)を開始するため、米国についてFDAにINDの申請を行いました。本剤は既にキメリックス社による欧米における臨床試験においてBCV Oralが高活性の抗ウイルス効果を示し、また広域のスペクトラムを有することが確認されており、各種dsDNAウイルスに対する幅広い抗ウイルス活性は、BCV IVに関しても造血幹細胞移植後の各種ウイルス感染症の予防及び治療に対する有効性と安全性が期待されます。
キメリックス社は、2020年12月、米国食品医薬品局(FDA)が天然痘の医学的防衛策としてBCVの新薬申請(NDA)の提出を受理したことを発表しました。FDAは優先審査を認め、処方薬ユーザー・フィー法(PDUFA)に基づき、審査終了目標日(PDUFA Date)を2021年月7日に設定しました。
(注17)ブリンシドフォビル(BCV)は、シドフォビル(CDV、欧米では既承認・販売の抗ウイルス薬、本邦は未承認)に脂肪鎖(ヘキサデシルオキシプロピル:HDP)が結合した構造となっており、速やかに脂質二重膜へ取り込まれ効率よく細胞内へ移行した後、細胞内ホスフォリパーゼによる代謝によって脂肪鎖が切り離され、生成された活性化体(CDV-PP:CDV diphosphate)が細胞内で長時間保持される結果、抗ウイルス活性が飛躍的に向上した化合物です。また、HDP結合により、OAT-1トランスポーターによる腎尿細管上皮細胞への蓄積が生じないことに加え、CDVが血中に遊離するレベルは低いため、CDVの根本的問題であった腎毒性を回避できます。
(注18) dsDNAウイルス:CMV、AdV、HHV-6、BKウイルス、HSV1/2、VZV、HPV、JCV、天然痘ウイルスなど、ヘルペスウイルス科、アデノウイルス科、ポリオーマウイルス科、パピローマウイルス科、ポックスウイルス科を含む。
(参考) 医薬品研究開発の一般的な進行について
医薬品研究開発のプロセスは以下のとおりであり、通常、(a)から(f)までに10年から17年程度かかるといわれています。
(医薬品研究開発のプロセス)
(a) 基礎研究
(b) 前臨床試験(非臨床試験)
(c) 臨床試験(治験)
(d) 申請及び承認
(e) 薬価申請・収載
(f) 上市販売
(g) 製造販売後調査
(a) 基礎研究
新薬のもとになる候補物質を探し出すプロセスです。化学物質、微生物、遺伝子などの研究から、将来薬となる可能性がある新しい物質(成分)を発見したり、化学的に作り出したりするための研究であり、一般的には研究所などで実施されます。
(b) 前臨床試験(非臨床試験)
(a)で特定された薬剤候補化合物を対象に、生物学的試験として、動物や培養細胞を用いて安全性や有効性について調べる、いわゆる動物に対して実施する試験です。また、化学的試験として、製造方法、原薬及び製剤の規格・安定性を調べるなどの試験があります。
(c) 臨床試験(治験)
前臨床試験の結果、有効性及び安全性の観点から有用な医薬品になり得る可能性が認められた場合、十分な検討の上で、実際にヒトを対象とした有効性及び安全性の検証を行う、臨床試験(治験)が行われます。治験はさらに3段階にわかれ、それぞれ参加者の同意を得た上で行われますが、その内容は以下のとおりです。
① 第Ⅰ相臨床試験
第Ⅰ相は、治療効果を見ることを目的とせず、比較的少数の健康な志願者を対象に主に副作用と安全性を確認する試験です。
② 第Ⅱ相臨床試験
第Ⅱ相は、通常、患者さんにおける治療効果の探索を主な目的とする試験を開始する段階です。少数の患者さんを対象に、有効性と安全な投薬量や投薬方法を確認する試験です。
③ 第Ⅲ相臨床試験
第Ⅲ相は、第Ⅱ相よりも投与患者数をさらに増やし、治療効果の既存薬剤との比較データ、副作用のデータ等を収集することによって、有効性と安全性について検証し、新薬として承認されるための適切な根拠となるデータを得ることを目的とした試験です。
(d) 申請及び承認
治験で有効性や安全性などが証明された治験薬について、新薬承認申請書類を作成し、厚生労働省に製造販売承認の申請を行います。数段階の審査を受け、承認されて初めて「薬」として市場に出ることになります。ちなみに基礎研究段階で新薬候補とされた物質(成分)の内、製造販売承認を得ることができるものはわずか2万分の1から2万5千分の1といわれております。
(e) 薬価申請・収載
新薬の価格(以下「薬価」といいます)を厚生労働省へ申請し、開発コスト、類似薬や諸外国の価格を参考に価格の承認を受けます。これを薬価収載といいます。
(f) 上市販売
薬価収載が完了し、実際に薬を販売できる状況になることを上市といい、この段階から販売が可能になります。
(g) 製造販売後調査
販売を開始した後に、病院などの医療機関でさらに多くの患者さんに投与された結果を元に、臨床開発段階では発見できなかった副作用や適正使用情報などの収集が行われ、厚生労働省に報告を行います。
(1) 当社の概要
当社は、元米国アムジェン社(注1)本社副社長で、同社の日本法人であるアムジェン株式会社(現在は武田薬品工業株式会社が全事業を譲受)の創業期から約12年間社長を務めた吉田文紀が、2005年3月に設立した医薬品企業です。
経営理念は「共創・共生」(共に創り、共に生きる)で表され、患者さんを中心として医師、科学者、行政、資本提供者を「共創・共生」の経営理念で結び、アンメット・メディカル・ニーズ(Unmet Medical Needs)(注2)に応えていくことにより、社会的責任及び経営責任を果たすことを事業目的としています。
なお、当社の事業は、医薬品等の研究開発及び製造販売並びにこれらの付随業務の単一セグメントであるため、セグメント別の記載を省略しています。
(注1) バイオ医薬品業界最大手。1980年、米国カリフォルニア州サウザンド・オークスにおいて、AMGen(AppliedMolecular Genetics)として設立。日本においては、1993年5月1日にアムジェン株式会社として業務を開始しました。なお、2008年2月に武田薬品工業株式会社がアムジェン株式会社の株式を100%取得後、現在は武田薬品工業株式会社が全事業を譲受しています。
(注2) アンメット・メディカル・ニーズ(Unmet Medical Needs)とは、未だ満たされない医療上の必要性を意味し、患者さんや医師から強く望まれているにもかかわらず有効な既存薬や治療がない状態を指します。
(2) 当社の事業の特徴
がん及び血液領域における希少疾病分野(注3)の研究開発の多くは、欧米を中心に、大手製薬企業よりもむしろ、多くの大学・研究所、バイオベンチャー企業により創薬研究・新薬開発が活発に行われ、海外では既に数々の有用な新薬が医療の現場に提供されています。しかし、これらの分野は開発に高度の専門性が求められることから、開発の難度も高く、また大手の製薬企業が事業効率の面、採算面で着手しにくいため日本を初めとするアジア諸国においては手掛けられていない空白の治療領域となっています。当社は、極めて医療上のニーズは高いものの、新薬の開発が遅れている空白の治療領域をビジネスチャンスと捉え、特に、高い専門性が求められ難度が高いために参入障壁の高いがん及び血液領域を中心とした日本初のスペシャリティ・ファーマ(注4)です。当社は、大型新薬(いわゆる売上高が1,000億円を超える「ブロックバスター」)の追求ではなく、マーケットは相対的に小規模でも医療ニーズの高い希少疾病分野を中心とした新薬開発に取り組み、これらの医薬品及び新薬候補品を数多く保有することにより、強固なパイプライン・ポートフォリオを構築し、高付加価値で高収益を達成し、持続性のある事業展開を行います。
当社は、このような空白の治療領域を埋めるための新薬の開発・提供を行うことを企業使命として設立されました。新薬が開発されないことで治療上の問題を抱えている患者さんに対して、短期間で開発をし、迅速に治療薬をお届けすることを最優先に考え、医療への貢献、そして医薬品業界の健全な発展に寄与することにより、持続的成長と安定への道を進んでまいります。
(注3) 希少疾病分野とは、患者数が少ない疾病分野のことで、この分野に対する医薬品は希少疾病用医薬品(Orphan Drug:オーファンドラッグ)と呼ばれます。厚生労働省はオーファンドラッグ制度を設定し、我が国において患者数が5万人未満の重篤な疾病であること、医療上特にその必要性が高いこと等をその指定の基準としています。当該指定を受けると、申請から承認までの期間が短縮され、再審査期間が最長10年になる等の優遇措置があります。
(注4) スペシャリティ・ファーマとは、得意分野において国際的にも一定の評価を得る研究開発力を有する新薬開発企業をいいます(2007年「新医薬品産業ビジョン」(厚生労働省)の定義による)。
(3) 当社の事業モデルについて
創薬系事業の特徴として、新薬の開発は長期間にわたり膨大な先行投資を強いられるものの、その研究開発の成功確率は極めて低いことが知られています。一般に、研究所において何らかの生物・生理活性(注5)が認められた化合物が新薬として承認にいたる確率は、2万分の1~2万5千分の1と言われています。また、承認を取得した新薬のうち、上市・販売後において採算が取れるのはそのうちの15~20%以下と言われています。当社は、このような創薬系事業の難しさを踏まえた事業モデルを構築しています。
当社では、開発にかかる様々なリスクと費用を軽減するとともに、開発候補品の臨床試験を迅速・確実に進め、開始から承認取得までの期間を短縮するために、主として既にヒトでPOC(Proof Of Concept)(注6)が確立され、前臨床試験データと臨床試験データがある化合物を対象としております。これらの化合物の探索は当社独自の探索ネットワークと評価ノウハウを活用して、社内の経験を有した専門スタッフによる第1次スクリーニングにより絞り込みを最初に行います。その後、科学的諮問委員会(Scientific Advisory Board:以下「SAB」といいます)(注7)において、第一線で関連分野における治療の研究に携わる経験豊かな社外専門家の厳密な評価を受けた上で、当社において最終的な導入候補品を決定いたします。
社内外の専門家による、こうした“目利き”のプロセスを経て、当社はがん及び血液領域を中心として、製薬企業、バイオベンチャー企業等から主にヒトでPOCが確立された開発品の日本並びにアジア諸国、さらには欧米を含むグローバルの開発・製造・販売権を継続的に確保することにより、持続性のある事業を展開しています。そのような、開発の成功確率が高く、事業性のある、魅力的な開発候補品を導入するためには、この“目利き”の力に加え、がん及び血液という開発の難度が高い治療領域における当社の開発力について、開発候補品の提供者であるライセンサーから高い評価を得ることも導入の成否を決める重要なポイントとなります。そのためには、①適切な治験計画の策定、②治療対象となる適切な治験患者の選定、③その領域における医学専門家と公正な関係を維持・構築できる、専門性の高い優秀な開発スタッフが必要となります。これらの総和が開発力となり、開発を着実に、かつ迅速に実行することが可能となります。がん及び血液分野で実績のある大手製薬企業の開発部門で経験を積んだ人材を中心に構築された当社の開発チームが導入から承認申請までを僅か4年間という短期間でなし得た、抗がん剤 SyB L-0501での実績は、ライセンサー、パートナー企業、導入候補先企業から高い評価を得ています。
なお、開発につきましては、基本的な開発戦略の中枢となる臨床試験のデザイン、海外の試験との連携、医学専門家との調整等は当社が主体となって手掛け、定型的な開発業務は、外部資源であるCRO(Contract Research Organization 受託臨床試験実施機関)(注8)へ業務委託し、製造についてはライセンス供給元あるいは信頼できる国内外の製薬企業へ業務委託を行います。
販売につきましては、2008年8月に締結した事業提携契約に基づき、エーザイ株式会社(以下「エーザイ」という)を通じて国内販売を行ってまいりました。事業提携契約が2020年12月に満了となることから、2018年10月よりトレアキシン®の国内販売について自社による販売体制構築の準備を開始しました。2020年12月からの自社販売体制への移行に向けて、がん及び血液領域に精通した自社MR(Medical Representative)(注9)を中核とした全国営業体制の構築と流通及び物流機能の整備を推進すると同時に営業戦略・企画の策定及び市場調査を行うマーケティング体制の強化に努めるとともに関係治療領域におけるKOL(Key Opinion Leader)(注10)との良好な関係構築、的確な医療ニーズの把握と市場調査を行い、各種データ、ノウハウの蓄積を図ってまいり、2020年12月の契約満了に伴い自社販売体制へ移行しました。
これらの事業モデルを図示すると以下のようになります。
(注5) 生理活性とは、化学物質が生体の特定の生理的調節機能に対して作用する性質のことです。この生理活性の作用を持つ化学物質を疾病治療に応用したものが医薬品となります。
(注6) POC(Proof of Concept)とは、新薬候補物質の有効性や安全性を臨床で確認し、そのコンセプトの妥当性を検証することを意味します。
(注7) 科学的諮問委員会(SAB:Scientific Advisory Board)とは、世界中から集まる膨大な新薬候補を元に、医療ニーズの高さや収益性などリスクバランスのとれたポートフォリオを、それぞれの専門の立場から意見や提言を交え徹底的に議論した上で、パイプライン戦略を構築する、当社の重要な評価機関です。当社では、SABを年2~3回開催し、世界中から優れた実績と経験をもつ臨床医・基礎科学者の方々に、当社の創薬研究及び新薬開発のアドバイザーとして参画いただいています。
(注8) CRO(Contract Research Organization)とは、製薬企業が、自社で実施する開発業務を遅滞なく進めるために、一部の業務について委託を行う機関です。委託業務の内容としては、治験が実施計画書どおりに遂行されているかをモニタリングするモニター業務や、臨床データを管理するデータ管理業務などがあります。
(注9) MR(Medical Representative)とは、自社医薬品に関する情報の専門家として医療機関を訪問し、医療関係者と面談することにより、医薬品の品質・有効性・安全性等に関する情報の提供・収集・伝達を主な業務とする医療情報担当者をいいます。
(注10)KOL (Key Opinion Leader)とは、担当領域の治療において他の医師に影響力を持つ医師のことをいいます。
(4) 当社の事業戦略
当社は、上記の事業を成功させるために、主に以下の5つの事業戦略を展開しています。
(a) ポストPOC戦略による開発リスクの軽
当社の導入候補品(注11)は、主として既にヒトでPOCが確認されていることを原則としています。従って、臨床開発ステージが比較的後期段階にある候補品か、既に海外で上市されている製品が対象となります。これらの導入候補品は既に海外で先行して開発が行われており、新薬としてヒトでの有効性・安全性が確認されていることから、開発リスクを軽減でき、また、先行している海外の治験データを活用することにより日本を含めアジアにおける開発期間を短縮するとともに開発コストを低減し、成功確率を高めることが可能となります。
(注11)導入候補品とは、当社の開発候補品として他社より開発権等の権利取得を検討している化合物を指します。
(注12)ブリッジングスタディとは、外国での臨床データを活用するために国内で行われる試験のことをいいます。この国内試験の結果を外国のデータと比較し、同様の傾向があることを確認します。
(b) 高度な探索及び評価能力による、優れたパイプラインの構築
当社の新薬サーチエンジンは、製薬企業及びバイオベンチャー企業等との多様なネットワークによって構築され、膨大な化合物の中から、社内の専門家による厳正な評価を経て、有望な導入候補品が抽出されます。これらの導入候補品は更に、第一線で研究に携わる経験豊かな専門家により構成されるSABに諮られ、そのアドバイスと評価を受けた上で導入候補品を決定しています。この開発品導入決定までの高度なスクリーニングプロセスは、既に海外において有効性・安全性が確認された開発品を導入するポストPOC戦略と相まって開発リスクの軽減と開発期間の短縮につながることになり、また、候補品が医療の現場において求められるものかどうかの医療ニーズの充足度に対する理解、及び上市後の収益予測の精度向上に貢献しています。
<当社の開発品導入プロセス>
(注13)CDAとは、Confidential Disclosure Agreementの略で、秘密保持契約書のことを意味します。
(c) ラボレス・ファブレス戦略による固定費抑制
当社は、一切の研究設備や生産設備を保有していません。研究設備・生産設備はともに固定費発生源の代表格ですが、当社はこれらを一切保有せずに、開発候補品の探索及び導入後は、開発品の開発戦略策定と実行等の付加価値の高い業務に専念し、そのほかに必要とされる定型的な開発業務は外注しています。これにより低コストの医薬品開発を実現するとともに、財務戦略の機動性を確保しています。
(d) ブルーオーシャン戦略(注14)による高い事業効率の実現
海外で標準治療薬として使用されている製品が日本では使用できない、あるいは海外で新薬として承認された製品が5年近くも遅れて日本で承認される、いわゆるドラッグ・ラグの問題が深刻化しており、がん患者の難民という言葉も生まれています。このドラッグ・ラグは、当社の戦略的開発領域である難治性のがん及び血液疾患領域で特に目立っています。特に抗がん剤の市場自体は大きく、また高齢化に伴い現在も拡大傾向にあるものの、抗がん剤の対象疾患は多岐にわたり、がん腫により細分化されているため、各々のがん腫でみると対象患者数がそう多くはない治療領域が数多く存在します。これらの領域での新薬の開発には、極めて高い専門性が求められ、開発の難度が高い半面、大手の製薬企業では採算性などの問題から開発に着手しにくいことがその理由のひとつといわれています。しかし、ひとたび、そうした領域において新薬の承認を取得し上市できれば、競合が少ないため、これらの領域で適応拡大・新製品上市を着実に積み上げていくことで、高成長・高収益を実現できるものと考えています。
(注14)ブルーオーシャン戦略とは、競合との熾烈な競争により限られたパイを奪い合う市場(レッドオーシャン)を避け、市場を再定義し、競合のいない未開拓な市場(ブルーオーシャン)を創造することで、顧客に高付加価値を与えつつ利潤の最大化を目指す戦略です。
(e) アジアからグローバル展開へ
当社はこれまで日本を中心としたアジア各国を対象に事業を展開してまいりました。しかしながら、日本の医療を取り巻く環境が大きく変わっていくなか、アジアに留まっていては大きな発展は望めません。今後はグローバルな展開を視野に入れた開発候補品の探索及び評価を実施してまいります。2019年9月30日にはキメリックス・インク社(本社:米国ノースカロライナ州、以下「キメリックス社」という)との間で抗ウイルス感染症治療薬ブリンシドフォビルに関しての独占的グローバルライセンス契約を締結し、当社は天然痘疾患を除くすべての疾患を対象とした世界全域における開発・販売に加えて製造を含む独占的権利を取得しました。
当社は、従来の日本への候補品の導入に限定せず、グローバルな市場への展開を目指して、開発に着手しております。抗ウイルス感染症治療薬ブリンシドフォビルにつき、播種性アデノウイルス(AdV)感染症及び免疫不全状態でのアデノウイルス(AdV)感染症を対象に、2021年3月10日、米国及び英国を中心とした国際共同第Ⅱ相臨床試験(フェーズ2a)を開始するため、米国についてFDAにINDの申請を行いました。
2.当社のパイプラインについて
当社は現在開発中のパイプラインとして、SyB L-0501、SyB L-1101、SyB C-1101、SyB L-1701及びSyB L-1702、SyB V-1901を有しています。今後も開発候補品を継続的に導入することにより、パイプラインのより一層の拡充及びリスク・リターンのバランスのとれたパイプライン・ポートフォリオを構築してまいります。
<当社パイプラインの進捗状況>
(1) [抗がん剤 SyB L-0501(凍結乾燥注射剤) / SyB L-1701(RTD製剤) / SyB L-1702(RI製剤))(一般名:ベンダムスチン塩酸塩またはベンダムスチン塩酸塩水和物、製品名:トレアキシン®)]
SyB L-0501の主成分であるベンダムスチン塩酸塩(一般名)は、ドイツにおいて非ホジキンリンパ腫(注15)、多発性骨髄腫及び慢性リンパ性白血病の治療薬(商品名「リボムスチン®」)として長年使用されている抗がん剤です。この製品の導入の背景としては、再発・難治性の低悪性度非ホジキンリンパ腫及びマントル細胞リンパ腫の患者さんには、この分野には優れた薬剤がなく、まさしく当社の企業使命である、空白の治療領域を対象とした薬剤であること、また当社の強みである分野(血液がん)であることが導入の決め手となりました。
2018年7月には日本血液学会が発行した造血器腫瘍診療ガイドラインにトレアキシン®とリツキシマブの併用療法(BR療法)が新たに収載され、既承認(再発・難治性の非ホジキンリンパ腫(低悪性度NHL)及びマントル細胞リンパ腫(MCL)、未治療(初回治療)の低悪性度NHL及びMCL並びに慢性リンパ性白血病(CLL))のすべての適応症において、標準的治療の選択肢として推奨されることになりました。これにより名実ともにトレアキシン®が悪性リンパ腫における標準療法として位置づけられています。
既に承認を取得した適応症に続き、再発・難治性のびまん性大細胞型B細胞リンパ腫(r/r DLBCL)を対象とするBR療法による第Ⅲ相臨床試験については、試験成績の主要評価項目である奏効率において期待奏効率を上回る良好な結果が得られたことを基に、2020年5月に製造販売承認事項一部変更承認申請し、2021年3月に承認を取得しました。
2017年9月にイーグル社との間で日本における独占的ライセンス契約を締結したトレアキシン®液剤(RTD製剤及びRI製剤(注16))については、RTD製剤は2020年9月18日に製造販売承認を取得し、2021年1月より販売を開始しました。RI製剤につきましては現在、安全性に関する臨床試験を実施中で、今年度中に承認申請の予定です。RTD製剤は、従来の凍結乾燥注射剤に比べて、手動による煩雑な溶解作業が不要で、そのために要する時間を短縮することができ、医療従事者の負担を大幅に低減することが可能となります。また、RI製剤は、投与時間が、従来の凍結乾燥注射剤及びRTD製剤の60分に対して10分間と大幅に短縮されるため患者さんと医療従事者の負担を大幅に低減することが可能となることから大きな付加価値を提供することができます。更には、液剤の製剤ライセンスによる複数の特許保護を通じてトレアキシン®の製品寿命を2031年まで延長し、当社事業の成長基盤をより強固なものとすることが可能となります。
(注15) 非ホジキンリンパ腫とは、白血球の中のリンパ球ががん化した悪性腫瘍である悪性リンパ腫のうち、ホジキンリンパ腫以外の総称です。日本人の悪性リンパ腫では、大半を非ホジキンリンパ腫が占めています。
(注16) RTD製剤及びRI製剤は、従来の凍結乾燥注射剤(FD)とは異なり既に液化された製剤です。RTD製剤(Ready To Dilute)は調剤作業を大幅に低減し、さらに急速静注であるRI製剤(Rapid Infusion)により点滴時間を従来の60分間から10分間に短縮することにより、FD製剤に比べ患者さんの負担を大幅に軽減し、さらには医療従事者に大きな付加価値を提供することが可能になります。
(2) [抗がん剤 SyB L-1101(注射剤) / SyB C-1101(経口剤)(一般名:Rigosertib Sodium<リゴセルチブナトリウム>)]
リゴセルチブ注射剤については、導入元であるオンコノバ・セラピューティクス社(本社:米国ペンシルベニア州、以下「オンコノバ社」という)が、現在の標準治療である低メチル化剤による治療において効果が得られない、治療後に再発した、または低メチル化剤に不耐容性を示した高リスク骨髄異形成症候群(高リスクMDS)における全生存期間を主要評価項目として、国際共同第Ⅲ相臨床試験(INSPIRE試験)を実施しておりますが、2020年8月に医師選択療法との比較において主要評価項目を達成しなかったことを発表しました。当社は日本における臨床開発を担当しており、INSPIRE試験の追加解析から得られた知見を今後のリゴセルチブの開発に活用するための検討を進めてまいります。
トレアキシン及びリゴセルチブに関して、東京大学医科学研究所との共同研究等を通じて、両化合物あるいは他の既存薬との併用により新たな有用性を見出すとともに新規適応症の探索を行い、事業価値の最大化に努めます。
(3) [抗ウイルス薬 SyB V-1901(一般名:Brincidofovir<ブリンシドフォビル>)]
当社は2019年9月30日にキメリックス社との間で抗ウイルス薬ブリンシドフォビルの注射剤及び経口剤(SyB V-1901、以下各々「BCV IV」及び「BCV Oral」という)(注17)に関しての独占的グローバルライセンス契約を締結し、天然痘疾患を除くすべての疾患を対象としたBCVの世界全域における開発・販売に加えて製造を含む独占的権利をキメリックス社から取得しております。当社は、2020年2月に開催したグローバルアドバイザリーボードでの検討の結果、「空白の治療領域」でアンメット・メディカル・ニーズの高い造血幹細胞移植後のアデノウイルス(AdV)感染症を対象に、日本/アメリカ/ヨーロッパを中心としたBCV IVのグローバル開発を優先的に進めることを決定しております。そして、当該試験により得られた有効性と安全性に関する知見に基づき、造血幹細胞移植後の各種dsDNAウイルス(注18)感染症に対する効果を検討し、抗マルチウイルス感染症へ対象領域を拡大し、更には腎臓移植を含む臓器移植分野等の対象領域拡大の可能性を追求することで、市場の拡大とBCVの事業価値の最大化を目指してまいります。BCV液剤の小児のアデノウイルス感染症を対象とした試験開始に向けて、播種性アデノウイルス(AdV)感染症及び免疫不全状態でのアデノウイルス(AdV)感染症を対象に、2021年3月10日、米国及び英国を中心とした国際共同第Ⅱ相臨床試験(フェーズ2a)を開始するため、米国についてFDAにINDの申請を行いました。本剤は既にキメリックス社による欧米における臨床試験においてBCV Oralが高活性の抗ウイルス効果を示し、また広域のスペクトラムを有することが確認されており、各種dsDNAウイルスに対する幅広い抗ウイルス活性は、BCV IVに関しても造血幹細胞移植後の各種ウイルス感染症の予防及び治療に対する有効性と安全性が期待されます。
キメリックス社は、2020年12月、米国食品医薬品局(FDA)が天然痘の医学的防衛策としてBCVの新薬申請(NDA)の提出を受理したことを発表しました。FDAは優先審査を認め、処方薬ユーザー・フィー法(PDUFA)に基づき、審査終了目標日(PDUFA Date)を2021年月7日に設定しました。
(注17)ブリンシドフォビル(BCV)は、シドフォビル(CDV、欧米では既承認・販売の抗ウイルス薬、本邦は未承認)に脂肪鎖(ヘキサデシルオキシプロピル:HDP)が結合した構造となっており、速やかに脂質二重膜へ取り込まれ効率よく細胞内へ移行した後、細胞内ホスフォリパーゼによる代謝によって脂肪鎖が切り離され、生成された活性化体(CDV-PP:CDV diphosphate)が細胞内で長時間保持される結果、抗ウイルス活性が飛躍的に向上した化合物です。また、HDP結合により、OAT-1トランスポーターによる腎尿細管上皮細胞への蓄積が生じないことに加え、CDVが血中に遊離するレベルは低いため、CDVの根本的問題であった腎毒性を回避できます。
(注18) dsDNAウイルス:CMV、AdV、HHV-6、BKウイルス、HSV1/2、VZV、HPV、JCV、天然痘ウイルスなど、ヘルペスウイルス科、アデノウイルス科、ポリオーマウイルス科、パピローマウイルス科、ポックスウイルス科を含む。
(参考) 医薬品研究開発の一般的な進行について
医薬品研究開発のプロセスは以下のとおりであり、通常、(a)から(f)までに10年から17年程度かかるといわれています。
(医薬品研究開発のプロセス)
(a) 基礎研究
(b) 前臨床試験(非臨床試験)
(c) 臨床試験(治験)
(d) 申請及び承認
(e) 薬価申請・収載
(f) 上市販売
(g) 製造販売後調査
(a) 基礎研究
新薬のもとになる候補物質を探し出すプロセスです。化学物質、微生物、遺伝子などの研究から、将来薬となる可能性がある新しい物質(成分)を発見したり、化学的に作り出したりするための研究であり、一般的には研究所などで実施されます。
(b) 前臨床試験(非臨床試験)
(a)で特定された薬剤候補化合物を対象に、生物学的試験として、動物や培養細胞を用いて安全性や有効性について調べる、いわゆる動物に対して実施する試験です。また、化学的試験として、製造方法、原薬及び製剤の規格・安定性を調べるなどの試験があります。
(c) 臨床試験(治験)
前臨床試験の結果、有効性及び安全性の観点から有用な医薬品になり得る可能性が認められた場合、十分な検討の上で、実際にヒトを対象とした有効性及び安全性の検証を行う、臨床試験(治験)が行われます。治験はさらに3段階にわかれ、それぞれ参加者の同意を得た上で行われますが、その内容は以下のとおりです。
① 第Ⅰ相臨床試験
第Ⅰ相は、治療効果を見ることを目的とせず、比較的少数の健康な志願者を対象に主に副作用と安全性を確認する試験です。
② 第Ⅱ相臨床試験
第Ⅱ相は、通常、患者さんにおける治療効果の探索を主な目的とする試験を開始する段階です。少数の患者さんを対象に、有効性と安全な投薬量や投薬方法を確認する試験です。
③ 第Ⅲ相臨床試験
第Ⅲ相は、第Ⅱ相よりも投与患者数をさらに増やし、治療効果の既存薬剤との比較データ、副作用のデータ等を収集することによって、有効性と安全性について検証し、新薬として承認されるための適切な根拠となるデータを得ることを目的とした試験です。
(d) 申請及び承認
治験で有効性や安全性などが証明された治験薬について、新薬承認申請書類を作成し、厚生労働省に製造販売承認の申請を行います。数段階の審査を受け、承認されて初めて「薬」として市場に出ることになります。ちなみに基礎研究段階で新薬候補とされた物質(成分)の内、製造販売承認を得ることができるものはわずか2万分の1から2万5千分の1といわれております。
(e) 薬価申請・収載
新薬の価格(以下「薬価」といいます)を厚生労働省へ申請し、開発コスト、類似薬や諸外国の価格を参考に価格の承認を受けます。これを薬価収載といいます。
(f) 上市販売
薬価収載が完了し、実際に薬を販売できる状況になることを上市といい、この段階から販売が可能になります。
(g) 製造販売後調査
販売を開始した後に、病院などの医療機関でさらに多くの患者さんに投与された結果を元に、臨床開発段階では発見できなかった副作用や適正使用情報などの収集が行われ、厚生労働省に報告を行います。