訂正有価証券届出書(新規公開時)
事業内容
当社は、日本が誇る優れた技術をもって難治性疾患を罹患された方々に新しい治療法を提供するべく、化学物質の合成によって医薬品を作製する従来型の化合物医薬品(低分子医薬品)分野に加え、当社が中核的な事業領域と位置付けているiPS細胞に関連する技術を活用した再生医療等製品(以下「iPSC再生医薬品」という。)分野において、医薬品の研究開発を行っております。
なお、当社の事業セグメントは、医薬品事業のみの単一セグメントであります。
(1)事業の概要
(ア)化合物医薬品分野
①概要
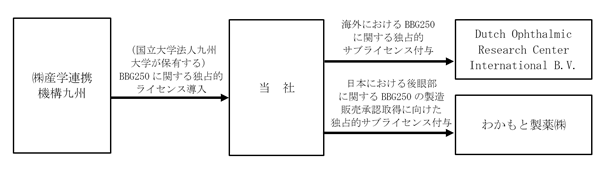
化合物医薬品分野では、国立大学法人九州大学(以下「九州大学」という。)の研究グループが発見したBBG250(Brilliant Blue G-250)という染色性の高い色素を主成分とした眼科手術補助剤を、株式会社産学連携機構九州からの独占的ライセンスに基づき開発しております。
当社は、上記の眼科手術補助剤に関する日本以外の全世界向けの独占的なサブライセンスをDutch Ophthalmic Research Center International B.V.(以下「DORC社」という。)に付与しており、DORC社は、平成22年9月からEU加盟国において、EU加盟国における安全規格に適合しているCEマーキング適合製品として、この眼科手術補助剤を製造・販売しております。この製品は、BBG250の高い染色性を利用して、眼内にある内境界膜又は水晶体を保護するカプセルを一時的に安全に染色し、硝子体・白内障の手術を行いやすくするものです。なお、CEマーキング適合製品とは、欧州における医療機器の製造販売のための必須要求事項を満たしたことを示すCE(フランス語におけるConformite Europeenneの略(英語におけるEuropean Conformity))マークの貼付を許諾された製品のことをいいます。
一方、日本国内については、わかもと製薬株式会社(以下「わかもと製薬」という。)に内境界膜を含む後眼部(網膜等、目の奥に当たる部分)についての独占的サブライセンスを付与しており、わかもと製薬が製造販売承認の取得に向けて開発を進めております。
なお、詳細については、後述「(3)化合物医薬品分野のパイプライン(開発コード:HLM0021、HLM0022、HLM0023)」をご参照ください。
化合物医薬品分野に係る事業系統図は以下のとおりであります。
 ②収益モデル
②収益モデル
当社は、上記図に記載のとおり、全世界で特許技術の実施許諾(サブライセンス付与)を行っております。このうち、欧州におけるサブライセンス先であるDORC社は、既にBBG250を使用した眼科手術補助剤を製造・販売しており、当社は、この売上に対してロイヤルティ収入を受け取っています。また、欧州以外の地域においてもサブライセンス先が開発を進めており、そのうち日本以外の地域においてはDORC社から承認を取得した後にロイヤルティ収入等を、日本においてはわかもと製薬からマイルストン収入及び承認を取得した後に製品の販売に応じた収入を得る計画となっています。なお、ロイヤルティ収入とは、契約に基づき、製品が上市された後に、その販売額に応じた一定料率を受領する収入をいい、マイルストン収入とは、契約に基づき、開発の進捗により予め定められた目標(マイルストン)の達成に応じて受領する一時的な収入をいいます。
(イ)iPSC再生医薬品分野
①概要
iPSC再生医薬品は、iPS細胞を分化誘導(細胞を特定の機能を持った細胞、例えば神経細胞・皮膚細胞などに人為的に変化させることをいう。)して作製した健康な細胞を移植することによって、高齢化などにより機能不全に陥った細胞等を置換して機能を回復することを目的としております。
当社では、国立研究開発法人理化学研究所(以下「理研」という。)の髙橋政代プロジェクトリーダー等が中心となって考案したiPS細胞由来の網膜色素上皮細胞(以下「RPE細胞」という。)への分化誘導方法等に関する知見を基にして開発した当社独自のノウハウを用いて、iPS細胞から分化誘導したRPE細胞の効率的な培養方法の確立に成功しました。
そこで、当社は、iPSC再生医薬品として製剤化されたRPE細胞(以下「RPE細胞製品」という。)を使って、根本的な治療法が確立されていない加齢黄斑変性の新たな治療法を開発すべく、より安全かつ効率的な生産方法の確立や治験に向けた準備を進めています。
以下、詳細なパイプラインの説明に先立ち、まず(i)iPS細胞、(ⅱ)RPE細胞と加齢黄斑変性及び(ⅲ)RPE細胞移植による加齢黄斑変性治療法に関してご説明いたします。
(ⅰ)iPS細胞
iPS細胞(人工多能性幹細胞:Induced Pluripotent Stem Cell)とは、平成18年に国立大学法人京都大学(以下「京都大学」という。)の山中伸弥教授が世界で初めて作製に成功し、平成24年にその功績からノーベル生理学・医学賞を受賞したことで広く知られるようになった、皮膚などの体細胞にいくつかの遺伝子(山中因子)を導入することによって作り出される、様々な組織や臓器の細胞に分化する能力(多能性)と、ほぼ無限に増殖する能力(増殖能)を持った細胞であります。
ヒトの体は約60兆個の細胞からなりますが、それらの細胞は全て元々一つの細胞であった受精卵が細胞分裂を繰り返し、それぞれ臓器・器官等を構成する細胞へと分化したものであります。受精卵が特定の細胞に分化していく流れは一方通行であり、従来の技術では一度分化した細胞を分化する前の細胞(幹細胞)に戻すことはできませんでした。ところが、皮膚細胞などの成熟した細胞にいくつかの遺伝子を導入することにより、新たに様々な細胞に分化する能力(多能性)とほぼ無限に増殖する能力(増殖能)を持たせることに成功したものがiPS細胞であります。
当社が開発するiPSC再生医薬品は、このような特徴を有するiPS細胞を特定の細胞に分化誘導し大量培養したうえで、医薬品として製剤化し、人体に移植することで、老化等の原因により機能不全に陥った細胞を健康な細胞に置換し、機能回復を図るものであります。当社は、iPSC再生医薬品の開発においては、当社の強みである①細胞培養、②製薬工業、及び③移植医療という3つの領域の知見が必要となると考えております。
iPSC再生医薬品のように人体の中にある物質を医薬品化した医薬品としては、例えば免疫機能を担っている抗体を用いた抗体医薬品などがありますが、このような医薬品はバイオ医薬品と呼ばれます。当社が開発を進めているiPSC再生医薬品も、生命の最小単位である細胞を用いたバイオ医薬品として、iPS細胞から十分に分化誘導して作製した健康な細胞を移植することによって、臓器移植に近似する治療効果が期待できる点に従来の医薬品と大きく異なる特徴を有しております。
なお、iPS細胞のような多能性幹細胞は、いずれも自然に特定の細胞に分化していく訳ではないため、特定の細胞に分化を誘導するためにはiPS細胞の作製とは別の技術が必要となります。
当社は、この点について、理研の髙橋政代プロジェクトリーダー等が考案したRPE細胞への分化誘導方法及び分化誘導されたRPE細胞等に関する知見を基に、独自の研究開発の結果としてRPE細胞の分化誘導方法及び効率的な培養方法に関するノウハウを蓄積しており、主としてiPS細胞から分化誘導されたRPE細胞をiPSC再生医薬品として提供することによる新しい治療法の実用化についてさらなる研究開発を行っております。
加えて、近年、細胞医薬品分野においては、罹患者自身から採取した細胞(自家細胞)由来の幹細胞を用いたもののみならず、安全性が確認された他人の細胞(他家細胞)由来の幹細胞を活用した医薬品などの研究開発が進んでおります。
当社は、このような流れに沿うものとして、RPE細胞へ分化誘導するために使用するiPS細胞に関して、国内においては、罹患者自身から採取した細胞(自家細胞)から作製されたもの(自家細胞由来iPS細胞)も選択肢には入れつつも、京都大学iPS細胞研究所から提供された他人の細胞(他家細胞)から作製されたもの(他家細胞由来iPS細胞)を用いることを前提として考えております。
なお、iPS細胞と比較される多能性幹細胞としてES細胞(胚性幹細胞:Embryonic Stem Cells。以下「ES細胞」という。)があります。ES細胞は、動物の受精卵から採取した多能性細胞で、同じく多能性と増殖能を備えているものの、受精卵から細胞を採取するという点の倫理的な問題等から国内では過去ヒトに関して研究開発が必ずしも十分に進んでこなかった歴史があります。
(ⅱ) RPE細胞と加齢黄斑変性
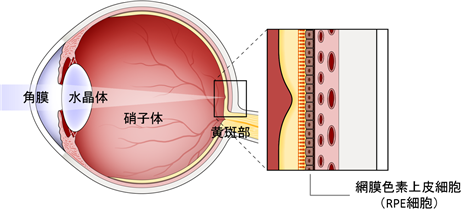
網膜は、光や色を感じる視細胞を含む感覚網膜(神経性網膜)と、RPE細胞と呼ばれる組織から構成されます。RPE細胞は、網膜の外側にある一層の細胞で、感覚網膜への栄養補給や老廃物の分解を担っています。そのため、RPE細胞の機能が低下すると視機能を担う感覚網膜の機能も低下してしまいます。
当社が最初の適応症(治療法の対象となる症状をいう。以下同じ。)として治療法の実用化に取り組んでいる加齢黄斑変性(AMD :Age-related Macular Degeneration)は、網膜変性疾患の一種であり、網膜の中でも視力を保つために極めて重要な役割を果たす「黄斑部」に障害が生じる病気で、発症すると次第に視力が低下したり、見え方に異常が生じるなどの症状が現われます。
(黄斑部と網膜色素上皮細胞)
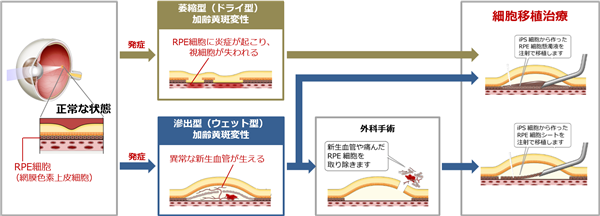
加齢黄斑変性は、滲出型(新生血管型:ウェット型)と非滲出型(萎縮型:ドライ型)に大別され、その原因は、黄斑部を支えるRPE細胞が老化等の原因により感覚網膜への栄養補給や老廃物の分解ができなくなってしまうことにあるものとされております。
日本人に多いウェット型は、黄斑部を支えるRPE細胞の機能不全に伴い、RPE細胞内に貯まった老廃物を分解するために、その外周にある脈絡膜から、脈絡膜新生血管と呼ばれる異常な血管が生えてくるのが特徴であります。この血管は正常な血管とは異なり、もろくて透過性が高いため、破れて出血し、又は水がしみだしてしまうため、網膜が浮腫を起こし、黄斑部の機能が阻害され、視力の低下や視野の歪みなどを生じます。
これに対して、欧米人に多いドライ型は、RPE細胞が加齢により萎縮してしまうことにより、網膜に障害が生じて視力が徐々に低下していく病気であります。
加齢黄斑変性の詳しい発症原因は未だ解明されておらず、根本的な治療法も確立しておりません。加齢黄斑変性は、欧米のような先進国では成人の失明原因として最も多く、公益財団法人難病医学研究財団 難病情報センターのホームページの記載によると、日本での推定罹患者数は平成19年時点で69万人(但し、罹患者数を正確に把握できないため、平成19年に福岡県内の人口約1万人の久山町において行われた調査結果を日本の人口に換算した推定値)と推定されております。
また、米国国立眼病研究所(National Eye Institute)のホームページにおいて公開されている統計データによると、平成22年時点で米国において207万人いると推定される加齢黄斑変性の罹患者は、平成42年(2030年)には366万人に増加すると予測されております。
(ⅲ)RPE細胞移植による加齢黄斑変性治療法
加齢黄斑変性の罹患者に対するRPE細胞の移植治療法は、これまでも多くの移植手術が試されておりましたが、これまでは特に移植対象となる細胞の確保が難しいという点が大きな課題でした。
特に平成18年に英国において罹患者自身の眼から切除して作製したRPE細胞のシートを移植するという治験が行われましたが、一定の有効性が確認されたものの、移植に必要な細胞を眼内の健常な箇所から切除する際の侵襲が大きく一般化はしませんでした。
これに対して、理研の髙橋政代プロジェクトリーダー等は、iPS細胞からRPE細胞を作製するという試みに取り組み、平成25年8月から開始された臨床研究(ヒトを対象とした研究)の中で、平成26年9月に世界で初めて、加齢黄斑変性の罹患者自身の皮膚の細胞から作製したiPS細胞(自家細胞由来iPS細胞)からRPE細胞を分化誘導し、RPE細胞のシートを作製したうえで当該罹患者に移植しました。かかる臨床研究に関して、平成27年3月に行われた日本再生医療学会において、半年経過後もがんはできておらず、細胞シートはきちんと定着しており、問題となる副作用は起きていない、また、異常な血管(上記(ii)の脈絡膜新生血管)を抑える注射治療を行っても続いていた視力低下が移植後には止まっている、と安全性及び有効性を示唆する発表がなされております。
当社のRPE細胞の移植治療法は、このような理研の髙橋政代プロジェクトリーダー等が中心となって考案したiPS細胞からRPE細胞を分化誘導し移植する技術・知見を基礎として、量産化・品質の安定化等に向けた当社独自の技術・知見を加えて開発したものであり、罹患者自身ではない第三者の細胞から作製され、安全性等に関する基準を満たしたiPS細胞(他家細胞由来iPS細胞)から作製したRPE細胞を含む懸濁液(以下「iPS細胞由来RPE細胞懸濁液」という。なお、懸濁液とは液体中に固体粒子が分散しているものをいう。)を注入し、又はiPS細胞由来RPE細胞のシート(以下「iPS細胞由来RPE細胞シート」という。)を移植し、患部に定着させることにより感覚網膜への栄養補給や老廃物の分解機能を回復させ、視機能を改善させる治療法であります。
当社は、日本人に多いウェット型の加齢黄斑変性と欧米人に多いドライ型の加齢黄斑変性の両方を適応症として、この治療法の実用化を目指しております。なお、当社としては、RPE細胞の移植による治療法は、悪くなったRPE細胞を健康なRPE細胞に置換するものであることから、網膜に関する疾患全般に対して効果があるものと期待しております。
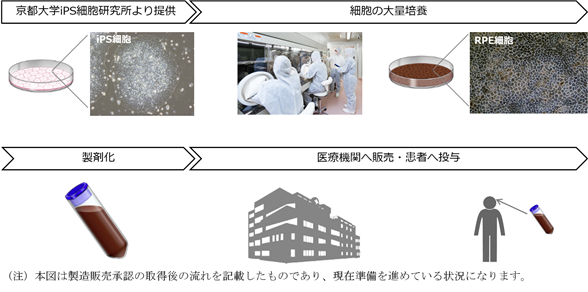
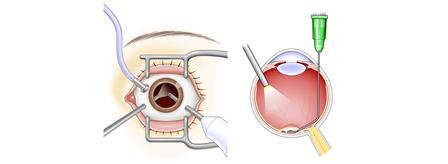
以下は、国内におけるiPS細胞の製造からRPE細胞製品の罹患者への投与までの流れを示す図であります。
また、以下は当社が実用化を目指しているiPS細胞由来RPE細胞懸濁液又はシートを用いた加齢黄斑変性の治療法を示す図であります。
② 開発に向けた事業上の取組み
当社は、平成25年2月にiPSアカデミアジャパン株式会社(以下「iPSアカデミアジャパン」という。)との間でRPE細胞を有効成分として含有する細胞製品を対象とする全世界を許諾領域としたiPS細胞樹立基本技術に関する特許実施権許諾契約を締結して非独占的ライセンスを受けるとともに、理研との間で同年3月にiPS細胞を含む多能性幹細胞由来RPE細胞を有効成分として含有する再生医療製品を対象とする全世界を許諾領域とした特許実施許諾契約を締結して独占的ライセンスを受けております。
また、当社は、かかるRPE細胞製品を用いた加齢黄斑変性の治療法の開発を迅速かつ確実に進めるべく、平成25年12月2日、大日本住友製薬株式会社(以下「大日本住友製薬」という。)との間で、日本におけるRPE細胞製品の開発を共同して行うことを合意し、同社との間で①当社の保有する知的財産権の実施許諾に関する実施許諾契約書(サブライセンス契約)、②共同開発を行う上での役割分担や費用負担を定めた共同開発契約書、並びに、③当該製品の製造や販売促進業務を受託する合弁会社の設立と同社への業務委託料等を定めた合弁契約書を締結いたしました。
これらの契約書のうち、実施許諾契約書においては、RPE細胞製品の日本における開発の進捗に伴って当社に支払われるマイルストン収入総額16億円(うち7億円は受領済み)について合意されており、また、共同開発契約書においては、当社がRPE細胞製品の開発に際して必要となる開発費用のうち最大52億円を大日本住友製薬が負担することが合意されております。即ち、当社がRPE細胞製品に関して、日本において平成26年11月25日に施行された医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律(以下「改正薬事法」という。)において再生医療等製品に関して新設された条件及び期限付承認(以下「条件付承認」という。)を取得し、これを販売するためには、本書提出日時点での合理的な見積もりとして平成32年までに、①細胞の特性解析試験・分析法開発・有効性確認試験等を含む前臨床試験や試薬等の消耗品に要する費用、②臨床試験と追加試験に要する費用、③人件費等の販管費などによって構成される開発費用などが必要になる見込みでありますが、当社は、これを当社の自己資金と大日本住友製薬からのマイルストン収入16億円及び同社による最大52億円の開発費用負担によって賄う予定であります。なお、連結損益計算書等における研究開発費の額は、大日本住友製薬による開発費用の負担分を控除した後の金額であります。
当社は、大日本住友製薬との間で、共同開発契約書に基づき、当社がRPE細胞製品の前臨床試験や臨床試験の実施、製造販売承認申請等を行うことに合意しております。他方、RPE細胞製品の製造や販売促進業務に関しては、大日本住友製薬が過去から培ってきた医薬品製造ノウハウや医薬品の販売網等を活かす形が望ましいと判断し、大日本住友製薬との合弁契約書に基づき、両社共同出資により平成26年2月28日付で設立された株式会社サイレジェン(以下「サイレジェン」という。)に対して、国内における製造及び販売促進業務を独占的に委託することに合意しております。
当社は、サイレジェンに対して、当社が考案した効率的な分化誘導方法の技術移管を進めており、承認取得後には、「再生医療等製品の製造管理及び品質管理の基準に関する省令」(以下「GCTP省令」という。なお、GCTPはGood Gene, Cellular, and Tissue-based Products Practiceの略)に従って、京都大学iPS細胞研究所から提供を受けたiPS細胞の親株からRPE細胞製品を製造する業務を委託することを予定しております。
当社は、このようにしてサイレジェンにおいて製造されたRPE細胞製品の独占的な供給を受けて、これを医療機関に販売することを予定しております。
なお、専ら開発に専念し製造・販売をライセンス先の製薬企業に任せるバイオベンチャーの一般的なビジネスモデルとは異なり、当社は、iPS細胞を用いた再生医療を新たな産業として捉え、単に研究開発に留まることなく、当社、サイレジェン及び提携企業によって、製造・販売に至るまでのバリューチェーンの全体を網羅できる体制の構築を目指しており、上述のとおり大日本住友製薬との間でサイレジェンを設立するのみならず、現在は技術者の手作業で行われている細胞培養の負担を軽減し、品質管理や効率化による原価低減の観点から重要と考えられる自動培養装置を、国立大学法人大阪大学(以下「大阪大学」という。)、株式会社ニコン(以下「ニコン」という。)及び澁谷工業株式会社(以下「澁谷工業」という。)との間で共同して開発しております。
さらに、当社は、RPE細胞製品の次のiPSC再生医薬品の候補を探索すべく、再生医療分野において実績を上げる大学・研究機関との共同研究を積極的に進めており、iPS細胞からヒト臓器を作る研究において実績を有する公立大学法人横浜市立大学(以下「横浜市立大学」という。)との間で、iPS細胞等を用いたヒト臓器の再生に関する基礎研究を既に開始しております。かかる基礎研究の内容については、「第2 事業の状況 3 対処すべき課題 (3)製品パイプラインの拡充について」をご参照ください。当社は、将来的に肝臓、腎臓及び膵臓といった臓器の再生も含め、アンメットメディカルニーズ(まだ有効な治療方法が存在していない疾患に対する治療方法開発の必要性)の高い領域についてパイプラインの拡充を模索してまいります。
iPSC再生医薬品分野に係る国内における事業系統図は以下のとおりであります。
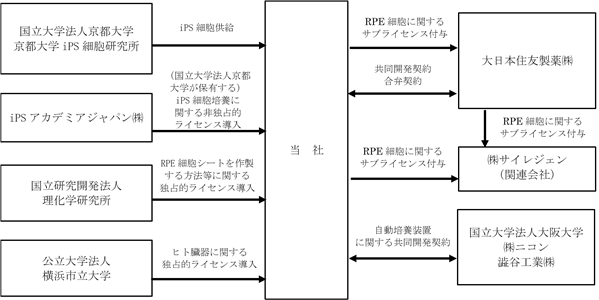 (注)平成26年2月に大日本住友製薬との合弁により設立したサイレジェンは、平成26年12月期においては持分法適用関連会社でしたが、平成27年12月期においては連結財務諸表の作成を予定していないため、本書提出日現在においては、持分法適用関連会社ではありません。
(注)平成26年2月に大日本住友製薬との合弁により設立したサイレジェンは、平成26年12月期においては持分法適用関連会社でしたが、平成27年12月期においては連結財務諸表の作成を予定していないため、本書提出日現在においては、持分法適用関連会社ではありません。
また、サイレジェンに対するRPE細胞に関するサブライセンス付与は、当社、大日本住友製薬及びサイレジェン間の共同実施許諾契約に基づき、当社及び大日本住友製薬から共同して行われております。
③ 収益モデル
当社は、国内では、共同開発先である大日本住友製薬からマイルストン収入を得るとともに同社による開発費用の負担を得ます。また、当社は、大日本住友製薬との合弁会社であるサイレジェンに製造及び販売促進業務を委託し、サイレジェンに対してこれらの委託費用を支払う一方で、サイレジェンの当社に対する製品売上に対してロイヤルティ収入を得るとともに、サイレジェンから供給を受けたRPE細胞を医療機関に販売することにより製品の販売収入を得る計画です。
収益モデルの概要は以下の図のとおりです。製品開発のリスクに関しましては、「第2 事業の状況 4 事業等のリスク」をご参照ください。また、契約の内容に関しましては「第2 事業の状況 5 経営上の重要な契約等」をご参照ください。
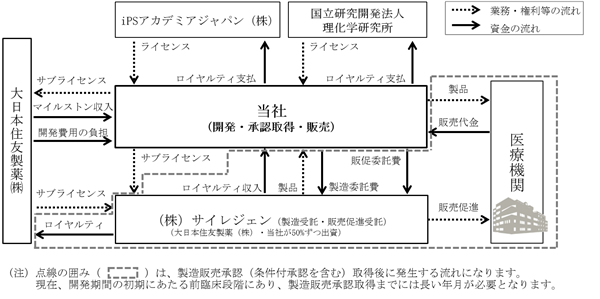 なお、当社が大日本住友製薬から得る予定のマイルストン収入は合計16億円(うち7億円は受領済み)であり、今後、国内における当該製品に関する治験計画届の提出から再生医療等製品に関して新設された条件及び期限付承認(以下「条件付承認」という。)の取得までの複数の目標(マイルストン)の達成により順次受領していく予定です。また、当社は、サイレジェンへの製造委託に基づき、当該製品の薬価の一定割合を製造単価として、サイレジェンから製品の供給を受けます。他方で、当社は、サイレジェンの当社への製品供給に関して、その正味売上高の額の一定割合(大日本住友製薬と同率)をロイヤルティ収入として受領します。さらに、当社は、販売促進業務をサイレジェンに委託し、当該製品の開発費用の総額と前年の年間総売上高の額に応じて変動する販売促進業務に係る業務委託料をサイレジェンに支払います。
なお、当社が大日本住友製薬から得る予定のマイルストン収入は合計16億円(うち7億円は受領済み)であり、今後、国内における当該製品に関する治験計画届の提出から再生医療等製品に関して新設された条件及び期限付承認(以下「条件付承認」という。)の取得までの複数の目標(マイルストン)の達成により順次受領していく予定です。また、当社は、サイレジェンへの製造委託に基づき、当該製品の薬価の一定割合を製造単価として、サイレジェンから製品の供給を受けます。他方で、当社は、サイレジェンの当社への製品供給に関して、その正味売上高の額の一定割合(大日本住友製薬と同率)をロイヤルティ収入として受領します。さらに、当社は、販売促進業務をサイレジェンに委託し、当該製品の開発費用の総額と前年の年間総売上高の額に応じて変動する販売促進業務に係る業務委託料をサイレジェンに支払います。
サイレジェンは、当社と大日本住友製薬が50%ずつの共同出資により設立した合弁会社であることから、サイレジェンの資本及び損益の50%分が実質的に当社に帰属いたします。
当社は、日本において条件付承認を取得した後に条件や期限を付されない承認(以下「本承認」という。)を取得し、また、欧米において製造販売承認を取得するための研究開発資金に充てるため、追加の資金調達を行うことを予定しております。当社は、増資等の方法により欧米における前臨床試験及び臨床試験を自社で継続し、RPE細胞製品の製造販売承認が得られた後に、医療機関への販売による収入を得るか、又は、欧米におけるRPE細胞製品の開発について共同開発先を探した上で米国における当該製品の開発に関する実施許諾契約(ライセンス契約)を締結し、契約締結及びマイルストン達成時のマイルストン収入、並びに上市後のロイヤルティ収入を得るか、のいずれかの方法により収入を得る計画です。
④ 当社の事業の特徴
以上のとおり、当社の事業は、とりわけ以下の点において特徴を有しております。
まず第一に、当社は、RPE細胞へ分化誘導するために使用するiPS細胞に関して、罹患者自身から採取した細胞(自家細胞)から作製されたもの(自家細胞由来iPS細胞)だけではなく、他人の細胞(他家細胞)から作製されたもの(他家細胞由来iPS細胞)を用いることを検討しております。国内においては、京都大学iPS細胞研究所より提供された他家細胞を用いる予定であります。
一般に自家細胞を使用する場合、罹患者が未知のウィルスに感染したり、免疫拒絶反応が起こるリスクは低いものと考えられます。しかしその一方で、罹患者ごとにiPS細胞の作製からRPE細胞の培養までを行う必要があるため、細胞調整に手間がかかり、罹患者個人間のばらつきが大きくなるほか、製造コストが膨らみ製造期間が長くなるなどの問題で多くの罹患者の方々を治療することが困難になると予想されます。さらに、眼内はそもそも免疫拒絶反応が起こりにくい免疫寛容の状態にあるとも言われており、他家細胞を使用する場合であっても、免疫拒絶反応が起こるリスクは高くないと考えられます。また、仮に免疫拒絶反応が起こるとした場合であっても免疫抑制剤を局所的に短期間使用することで対応することも可能と考えられます。
そこで、当社としては、免疫拒絶反応その他の安全面での課題に取り組みつつ、他家細胞から作製されたiPS細胞を用いたRPE細胞の移植による治療方法を確立させることを目指しております。
第二に、治療法実用化後の量産を可能とするため、当社は前述のとおり、iPS細胞の培養及びRPE細胞への分化誘導を担う自動培養装置を、大阪大学、ニコン及び澁谷工業との間で共同して開発しております。この自動培養装置により、現在は技術者の手作業で行われている細胞培養等の負担を軽減し、品質管理・効率化による原価低減を達成することで、上市後の当社製品の幅広い普及を図ることを検討しております。
第三に、当社は、製品の製造供給権を自ら保有し、自社で国内における製造販売承認の取得を目指していることが挙げられます。前述の通り、バイオベンチャーの一般的なビジネスモデルにおいては、自社は専ら開発に専念し、製造・販売をライセンス先の製薬企業に任せ、売上高に応じたロイヤルティ収入を得る例が多くみられます。しかしながら、この収益モデルの場合には一般的に製造・販売を行った製薬企業が利益の大半を獲得する場合が多いと考えられます。
これに対し、当社は、当社、サイレジェン及び提携企業によって、開発から承認取得、製造販売に至るまでのバリューチェーンの全体を網羅する体制を構築することを計画しております。当社は、かかる収益モデルによって、サイレジェンからのロイヤルティ収入に加え、製品の販売に伴う収入も得られる予定です。
(2)当社のパイプライン(製品開発群)
以下の表は、当社の開発品並びにその適応症、市場、開発段階及び本書提出日現在の進捗状況を示しております。
なお、製品の開発に際しては様々なリスクを伴います。当社製品の開発リスクの概要については、「第2 事業の状況 4 事業等のリスク」のとおりであります。
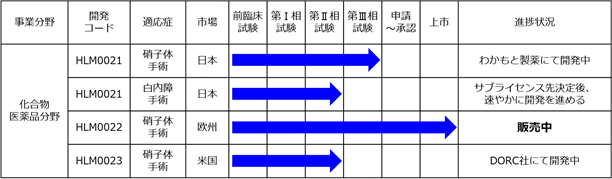
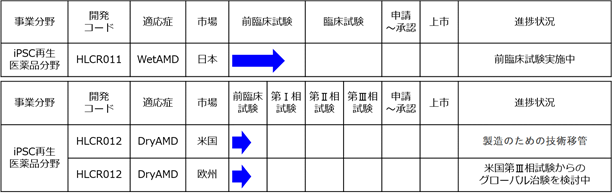 (注)1.「適応症」のうち、WetAMDとはウェット型の加齢黄斑変性をいい、DryAMDとはドライ型の加齢黄斑変性をいいます。
(注)1.「適応症」のうち、WetAMDとはウェット型の加齢黄斑変性をいい、DryAMDとはドライ型の加齢黄斑変性をいいます。
2.「前臨床試験」、「第Ⅰ相試験」、「第Ⅱ相試験」及び「第Ⅲ相試験」とは、改正薬事法に基づき、医薬品の製造販売承認を得るために必要となる試験の各段階を示すものであります(日本におけるiPS細胞由来RPE細胞移植による加齢黄斑変性治療法(HLCR011)は除きます。HLCR011に関しては、注4.をご参照ください。)。前臨床試験とは、臨床試験の前段階として行われる試験であります。第Ⅰ相試験とは、治験薬を少人数の健常者に投与して主として副作用などの安全性を検査する臨床試験です。第Ⅱ相試験とは、少数の罹患者を対象に治験薬の有効性と安全性を検査する臨床試験であり、基礎データを収集し、適応症や使用量等を決定するためのものです。第Ⅲ相試験とは、第Ⅱ相試験の結果を基に、より多くの罹患者を対象に治験薬の有効性と安全性を検査する臨床試験であり、より詳細なデータ収集を治験薬の使用条件等に反映するためのものです。第Ⅰ相試験は、健常者を対象とするものですが、侵襲性が高いなどの理由から最初から罹患者に対して行うべきと考えられる臨床試験に関しては、少人数の罹患者を対象に「第Ⅰ相/第Ⅱ相試験」として、第Ⅰ相試験と第Ⅱ相試験を同一の試験で実施する場合があります。なお、臨床試験とは、ヒトを対象として行われる研究(以下「臨床研究」という。)の中でも特に治療法等のヒトへの有効性・安全性を評価することを目的とするものをいい、特に新薬開発等を目的とした臨床試験を「治験」といいます。
3.「HLM0022」は、サブライセンス先のDORC社が製造販売を行っております。
4.「HLCR011」は、改正薬事法に基づき再生医療等製品に関して新設された条件付承認に関する規定に基づき条件付承認の取得を目指しております。従って、従来の医薬品のような開発の相(第Ⅰ相、第Ⅱ相、第Ⅲ相)の考え方は適用されません。条件付承認とは、再生医療等製品に関して、安全性が確認され、有効性が推定された場合に条件及び期限を付した上で製造販売承認を与える制度で、従来の医薬品のように有効性及び安全性の確認が必要な製造販売承認よりも早い時期に承認の取得が可能になると期待されている制度であります。条件付承認を得た場合には、再生医療等製品の適正な使用の確保のために必要な条件を遵守した上で期限まで製造販売を行うことができます。また、当該再生医療等製品の使用成績に関する資料等を収集した上で、当該期限内に再度製造販売承認の申請を行う必要があり、これにより有効性及び安全性が確認できた場合に改めて本承認が得られることになります。
5.「HLCR012」(米国向け)は、米国における製造販売承認を得るにあたって準拠すべき基準「cGMP」(Current Good Manufacturing Practiceの略)に則った製造のための技術移管に着手しております。
(3)化合物医薬品分野のパイプライン(開発コード:HLM0021、HLM0022、HLM0023)
HLM0021、HLM0022及びHLM0023は、内境界膜又は水晶体を保護するカプセルを一時的に安全に染色し、硝子体・白内障の手術を行いやすくする、BBG250(Brilliant Blue G-250)を主成分とする眼科手術補助剤であります。HLM0021、HLM0022及びHLM0023は、九州大学の研究グループが発見した染色性の高い色素BBG250を基に、当社の代表取締役社長である鍵本忠尚により平成17年に設立されたアキュメンバイオファーマ株式会社(現 アキュメン株式会社。以下「アキュメン」という。)が開発した眼科手術補助剤であり、当社は、平成25年12月20日付でアキュメンからBBG250を含有する眼科手術補助剤に関する事業を譲り受けております。当社は、かかる眼科手術補助剤に関する日本以外の全世界向けの独占的サブライセンスをDORC社に対して付与しており、これによりロイヤルティ収入を受け取っております。また、日本向けの内境界膜染色に関する開発、使用、販売について独占的サブライセンスをわかもと製薬に対して付与しており、これによりマイルストン収入及び製品の販売に応じた収入を得る計画となっております。
眼内は、硝子体というゼリー状の物質で満たされており、その奥に網膜がありますが、網膜剥離等の手術を行うためには、その前段階として、硝子体を切除し、網膜の最表層部分にあり網膜を固く伸ばす機能を有する内境界膜を剥離しなければなりません(このような硝子体の切除を伴う一連の手術を一般的に硝子体手術といいます。)。ところが、内境界膜は、非常に薄く透明な膜であるため、従来は手術経験が豊富な医師以外には剥離が難しいものでした。そこで、当社は、この内境界膜を一時的に青色に染色し、硝子体手術をより安全に行うことを可能にする染色剤としてBBG250を含有する眼科手術補助剤の開発を進めております。
また、白内障は、本来透明であるレンズが老化等の原因で白く濁り硬化することで、視力が低下する病気です。ヒトのレンズは柔らかく、透明な薄い膜でできたカプセルに入っているため、現行の白内障手術ではこの透明なカプセルの真ん中を丸く切り抜き、濁ったレンズの中身を人工の透明なレンズと入れ替えます。そこで、当社は、白内障に関しても、この透明なカプセルを一時的に青色に染色し、手術をより安全に行うことを可能にする染色剤として、同じく眼科手術補助剤の開発を進めております。
HLM0021、HLM0022及びHLM0023の開発方針等は、以下のとおりです。
① HLM0021(日本向け眼科手術補助剤(硝子体手術))
HLM0021(硝子体手術)は、現在日本での第Ⅲ相試験が終了し、製造販売承認の申請に向けて、原薬及び製剤の製造体制の確立、並びに、かかる原薬及び製剤での適切な安定性試験を実施する必要がある眼科手術補助剤(医薬品候補)であり、当社から日本における眼科手術用途(内境界膜染色に限る。)について独占的なサブライセンスを受けた、わかもと製薬により開発が行われております。当社は、治験用の原薬・製剤の供給や必要な試験の実施、製造委託先との調整等の業務を行い、わかもと製薬による内境界膜染色に関するHLM0021の製造販売承認の取得を支援してまいります。
HLM0021は、HLM0022(欧州向け眼科手術補助剤)のようなCEマーキング適合製品という区分ではなく、医薬品と区分されます。
日本において医薬品が製造販売承認を得るためには、(i)「治験薬の製造管理、品質管理等に関する基準」(以下「治験薬GMP」という。なお、GMPはGood Manufacturing Practiceの略)に基づく、ヒトに投与可能な治験薬の準備、(ii)「医薬品の臨床試験の実施の基準に関する省令」(以下「GCP省令」という。なお、GCPはGood Clinical Practiceの略)に基づく、新薬のヒトにおける有効性と安全性を確認するための臨床試験、並びに、(iii)「医薬品及び医薬部外品の製造管理及び品質管理の基準に関する省令」(以下「医薬品GMP」という。)に基づく、製造販売承認を得た後に医薬品の製造及び品質を適切に管理できる体制の具備といった要件を充足する必要があります。
もっとも、HLM0021の主成分は、平成22年に欧州のCEマーキング適合製品として有効性・安全性の確認を得て製造販売が行われているHLM0022の主成分と同じ化合物BBG250であります。そこで、(i)国内でHLM0022の製剤を用いて第Ⅲ相試験を実施すること、(ii)医薬品GMPに基づく原薬及び製剤を製造する体制を確立し、原薬及び製剤について安定性試験を実施すること、並びに、(iii)第Ⅲ相試験で得られた臨床試験の成績と今まで欧州で用いられた安全性に関する成績を比較した結果、医薬品GMPに基づき製造された製剤の品質がHLM0022の品質と同様であることについての適切な説明を申請資料中に記載することの3つの条件を満たすことによって製造販売承認の申請が行われる予定です。
このうち、HLM0022の製剤を用いた第Ⅲ相試験については、既に九州大学が中心となった医師主導治験が実施され、平成26年10月には、硝子体手術時の内境界膜の可視化に有効であり、手術の容易性が向上することが確認されるとともに、臨床的に重要な安全性の問題は認められなかった、との結果が得られております。また、当社は、医薬品GMPに基づく新たな原薬の合成法の技術開発を終え、原薬製造体制の確立に向けての検討を行っており、かかる製造体制が確立された後、原薬の安定性試験を実施する予定です。一方、製剤の開発に関しましては、前述の原薬を用いて、CEマーキング適合製品(HLM0022)を製造している受託製造会社において製剤の作製を行うにあたっての課題の抽出が終了し、製剤の製造に向けての準備が行われております。製剤に関しても製造体制が確立された後、原薬と同様に安定性試験が実施される予定であります。
以上から、今後、医薬品GMPに基づく原薬及び製剤の製造体制が確立され、原薬及び製剤について1年間分の安定性試験のデータが得られた時点で、安全性に関する成績の比較等を行うことで、製造販売承認の申請が可能になるものと考えております。
以上を前提に、当社としては、当社委託先による医薬品GMPに合致した原薬及び製剤の1年間分の安定性試験のデータが得られると見込まれる平成29年第1四半期頃に、製造販売承認申請が可能になると見込んでおります。但し、「第2 事業の状況 4 事業等のリスク」に記載のとおり、製品開発に際しては様々なリスクが伴うため、当社として上記の各予定時期を保証できるものではありません。また、実際の開発はわかもと製薬の判断によって行われることになります。このため、当社として現段階で製造販売承認申請の具体的な時期又はわかもと製薬による最終的な製造販売承認の取得を保証することはできません。
② HLM0021(日本向け眼科手術補助剤(白内障手術))
HLM0021(白内障手術)は、現在、当社が日本国内において白内障手術向けのサブライセンス先の選定を行っている眼科手術補助剤(医薬品候補)であります。当社は、かかる適応症に関しても今後サブライセンス先による治験、製造販売承認の取得を目指す予定であります。但し、現時点で具体的なサブライセンス先は決定しておらず、また、実際の開発はサブライセンス先の判断によって行われることになります。加えて、「第2 事業の状況 4 事業等のリスク」に記載のとおり、製品開発に際しては様々なリスクが伴います。このため、当社として現段階で今後の開発スケジュールを明言し、又はサブライセンス先による最終的な製造販売承認の取得を保証することはできません。
③ HLM0022(欧州向け眼科手術補助剤:商品名「ILM-Blue」)
HLM0022は、BBG250に関する日本を除く全世界向けの独占的なサブライセンスを付与しているDORC社により、欧州の必須安全要求事項(ESRs:Essential Safety Requirements)に適合したことを示すCEマーキング適合製品としてEU加盟国において平成22年9月から製造販売が行われている、硝子体手術向けの眼科手術補助剤であります。
④ HLM0023(米国向け眼科手術補助剤)
HLM0023は、BBG250に関する日本を除く全世界向けの独占的なサブライセンスを付与しているDORC社により開発が進められている米国向けの眼科手術補助剤(医薬品候補)であります。
HLM0023は、日本向けのHLM0021と同様に米国において医薬品と区分されますが、同国における医薬品の製造販売承認申請にあたっては、一般に、日本の医薬品GMPに対応する米国の基準(cGMP)に則った製剤を用いて治験を実施することが求められます。
開発を行うDORC社からは、米国の食品医薬品局(以下「FDA」という。なお、FDAはFood and Drug Administrationの略)に対する相談の結果、欧州におけるHLM0022の臨床成績を使用することが認められ、現在、第Ⅲ相試験に関する検討作業を行っていると聞いております。
もっとも、米国における第Ⅲ相試験の具体的な開始時期等及び製造販売承認の申請時期は、現在検討中の製造法が確定した時点で、実際の開発を行うDORC社によって最終的に決定されます。また、「第2 事業の状況 4 事業等のリスク」に記載のとおり、製品開発に際しては様々なリスクが伴います。このため、当社として現段階でこれらの具体的な予定時期を明言し、又はDORC社による最終的な製造販売承認の取得を保証することはできません。
(4)iPSC再生医薬品分野のパイプライン(開発コード:HLCR011、HLCR012)
HLCR011(日本向け)及びHLCR012(欧米向け)は、平成25年6月から日本において前臨床試験を開始し、治験に向けた準備を行っている再生医療等製品の候補であります。
当社は、平成25年2月にiPSアカデミアジャパンとの間でRPE細胞を有効成分として含有する細胞製品を対象とする全世界を許諾領域としたiPS細胞樹立基本技術に関する特許実施権許諾契約(非独占)を締結するとともに、同年3月に理研との間でiPS細胞を含む多能性幹細胞由来RPE細胞を有効成分として含有する再生医療製品を対象とする全世界を許諾領域とした特許実施許諾契約(独占)を締結しております。
当社は、HLCR011(日本向け)に関して、大日本住友製薬と平成25年12月2日付で実施許諾契約書、共同開発契約書及び合弁契約書を締結しており、共同開発先である大日本住友製薬から開発の進捗に応じてマイルストン収入合計16億円(うち7億円は受領済み)を得るほか、最大52億円までの開発費用の負担を得ることに合意しております。製造販売承認の取得後には、製剤化されたRPE細胞を医療機関に販売することにより医薬品販売収入を得るとともに、大日本住友製薬との共同出資による合弁会社であるサイレジェンに製造及び販売促進業務を委託し、同社から製品の当社への供給に関してロイヤルティ収入を得る計画です。
なお、当社は、HLCR011(日本向け)について条件付承認を取得した後に本承認を取得し、また、HLCR012(米国向け)及びHLCR012(欧州向け)の製造販売承認を取得するための研究開発資金に充てるため、追加の資金調達を行うことを予定しております。当社は、増資等の方法により欧米における前臨床試験及び臨床試験を自社で継続し、RPE細胞製品の製造販売承認が得られた後に、医療機関への販売による収入を得るか、又は、欧米におけるRPE細胞製品の開発について共同開発先と実施許諾契約(ライセンス契約)を締結しマイルストン収入及び上市後のロイヤルティ収入を得るか、のいずれかの方法により収入を得る計画です。
① HLCR011(日本向けiPS細胞由来RPE細胞懸濁液又はシート)
HLCR011は、iPS細胞を正常なRPE細胞に分化誘導し、純化した上で、iPS細胞由来RPE細胞懸濁液又はiPS細胞由来RPE細胞シートという形で罹患者に移植し、加齢黄斑変性の治療を行うiPSC再生医薬品候補であります。
iPS細胞由来RPE細胞の懸濁液の注射による治療法については、平成26年10月にES細胞由来RPE細胞を用いたAdvanced Cell Technology, Inc.(現 Ocata Therapeutics, Inc.)が実施した米国での臨床成績が発表されており、第Ⅰ相/第Ⅱ相試験にて良好な安全性が示され、有効性が推定されています。そこで、当社においてもiPS細胞由来RPE細胞懸濁液による治療法の開発を優先的に検討しております。
なお、当社のiPS細胞由来RPE細胞懸濁液による治療法は、理研の髙橋政代プロジェクトリーダー等が中心となって考案したiPS細胞からRPE細胞を分化誘導し移植する技術・知見を基礎として、量産化・品質の安定化等に向けた当社独自の技術・知見を加えて開発したものであり、細胞培養・分化誘導等の技術は基本的に同じであるものの、①罹患者自身ではない第三者の細胞から作製され、安全性等に関する基準を満たしたiPS細胞(他家細胞由来iPS細胞)を使用する点、②RPE細胞のシートではなく、RPE細胞を含む懸濁液を優先的に検討している点、③量産化及び条件付承認の取得のための基準への適合を目的とした工程変更が加えられている点等において異なるものであります。また、当社は、理研の髙橋政代プロジェクトリーダー等の臨床研究とは別個に改正薬事法に基づき厳格な基準に則った臨床試験(治験)を新たに実施しなければなりません。
HLCR011は、改正薬事法において再生医療等製品と区分されるものです。再生医療等製品は、化合物医薬品と異なり、数多くの細胞を培養した結果として作製された均質でない細胞を用いた医薬品であることから、全ての治験薬の成分が同質であることを前提とした従来の医薬品のための治験手続を経ることを求めた場合には、より多くの人数を対象とした臨床試験で安全性と有効性の確認が必要となり、開発の難易度が高まるとともに開発期間も長期に亘る上、これに伴うコスト増の結果、開発そのものに対するハードルが大幅に高くなります。そこで、かかる問題を是正するため、改正薬事法は、新たに再生医療等製品の性質により適した制度として条件付承認制度を設けており、この結果、再生医療等製品の製造販売承認の申請者は、(i)安全性・有効性を確認するに足りる十分な症例数の臨床試験を通じて行われる、従来の医薬品に準じた承認申請手続によって、条件や期限を付されない承認(以下「本承認」という。)を受けるプロセスのほか、(ii)より少人数を対象とした臨床試験を通じて、安全性の確認と有効性の推定を得ることにより、条件及び期限を付された条件付承認を受けるプロセスを活用することができるようになっております。
かかる条件付承認制度は、条件及び期限の設定により罹患者の安全に配慮しつつ、罹患者の新薬へのアクセスを早期に実現するものであり、かつ、申請者において罹患者に対する再生医療等製品の使用成績等の情報を蓄積し、安全性及び有効性を迅速に確認することを可能とするものであります。少ない症例数の臨床試験の試験成績から安全性の確認及び有効性の推定を得て条件付承認を受けた申請者は、かかる条件を遵守した上で期限まで再生医療等製品を実際に罹患者に使用し、以後その使用の成績等に関する調査を行った上で、期限内に改めて製造販売承認の申請をしなければならず、その結果、安全性の確認及び有効性の確認が得られれば条件及び期限の付されない本承認を受けることができます。
当社は、上記のような条件付承認制度を活用することを念頭に、再生医療等製品の候補としてのHLCR011について平成29年から第Ⅰ相/第Ⅱ相試験に相当する臨床試験を数十症例程度の規模で開始する予定であり、そのうち一部の罹患者に対する投与が完了し、投与後数か月分の試験成績の蓄積があった時点、即ち、最も早い場合で平成31年下期頃に製造販売に係る条件付承認の申請を行う予定です。そして、審査期間中に、残りの罹患者への投与後数か月分の試験成績を追加提出する予定です。但し、必要となる症例数、データ蓄積期間、及びこのような試験成績の追加提出を前提とする条件付承認の申請の可否については、現在何ら指針がなく、具体的なスケジュールと併せて、第Ⅰ相/第Ⅱ相試験の実施に先立ち、平成28年中を目途として医薬品や医療機器などの製造販売承認審査及び治験等に関する指導・助言を行う独立行政法人医薬品医療機器総合機構(以下「PMDA」という。なお、PMDAとはPharmaceutical and Medical Devices Agencyの略)と対面助言を行った上で確定される予定であります。
さらに、当社は、条件付承認を受けることができた場合、新たに市販後臨床試験を実施するのではなく、HLCR011を投与した罹患者に対する市販後調査を行い、その成績をもって本承認の取得に向けた製造販売承認の再申請を行うことができるか検討中ですが、これについては、平成28年中を目途とした上記のPMDAとの対面助言の中で併せて協議を行い、最終的には承認申請を行った後の承認審査の過程で確定される予定であります。
以上のとおり、当社は、既存の治験実務に従って目標とすべき各段階の予定時期を設定しておりますが、改正薬事法に基づく条件付承認制度を活用した治験手続に関しては未だ運用実績がないことから明らかでない点も多く、また、「第2 事業の状況 4 事業等のリスク」に記載のとおり、製品開発に際しては様々なリスクが伴うため、当社として上記各予定時期又は当該製品に関する製造販売承認の取得を保証できるものではありません。
なお、かかる治療法は、現在のところ前臨床試験を実施している段階にあり、未だ臨床試験も開始されておらず、ヒトに対する有効性及び安全性が確認されておりません。さらに条件付承認の条件が解除されるまでに現時点から10年以上の期間を要する可能性もあり、今後開発の失敗や遅延のリスクも低くはありません。
② HLCR012(米国・欧州向けiPS細胞由来RPE細胞懸濁液又はシート)
HLCR012は、ドライ型加齢黄斑変性を適応症としたiPS細胞由来RPE細胞懸濁液又はシートの移植による治療法であり、米国・欧州におけるiPSC再生医薬品候補であります。
米国・欧州においては、日本において平成26年11月25日に施行された改正薬事法に基づく条件付承認制度と同様の制度が存在しないため、従来の医薬品同様、第Ⅰ相試験から第Ⅲ相試験までの臨床試験を経て、各国の薬事法に基づく製造販売承認申請を行う必要があります。
当社は、調達資金を研究開発費に充当することによって、米国におけるRPE細胞製品に係る前臨床試験を継続する計画です。
このため、当社は、まずは米国において、医薬品受託製造会社(以下「CMO」という。)に対して、治験及び承認後の販売に使用されるiPS細胞由来RPE細胞の培養法及び品質管理試験方法についての技術移管を行っているところであります。
当社は、HLCR011(日本向け)について条件付承認を取得した後に本承認を取得し、また、HLCR012(米国向け)及びHLCR012(欧州向け)の製造販売承認を取得するための研究開発資金に充てるため、追加の資金調達を行うことを予定しております。なお、当社は、追加の資金調達の方法として、増資のほか、欧米におけるRPE細胞製品の開発について共同開発先を探したうえで実施許諾契約(ライセンス契約)を締結し、契約締結及びマイルストン達成時のマイルストン収入、並びに上市後のロイヤルティ収入を得ることなども柔軟に検討してまいります。
HLCR012(米国向け)の具体的なスケジュールについては、今後、臨床試験実施のための米国FDAとの事前会議等を通じて検討していく予定です。
また、欧州については、米国の第Ⅰ相/第Ⅱ相試験の結果を使って、第Ⅲ相試験から試験を実施することを検討しております。
なお、「第2 事業の状況 4 事業等のリスク」に記載のとおり、製品開発に際しては様々なリスクが伴うため、当社として上記各予定時期又は当該製品に関する製造販売承認の取得を保証できるものではありません。また、かかる治療法は、現在のところ前臨床試験を実施している段階にあり、未だ臨床試験も開始されておらず、ヒトに対する有効性及び安全性が確認されておりません。さらに欧米において承認を取得するまでに現時点から10年以上の期間を要する可能性もあり、今後開発の失敗や遅延のリスクも低くはありません。
なお、当社の事業セグメントは、医薬品事業のみの単一セグメントであります。
| 当社のiPSC再生医薬品分野における主要パイプラインであるHLCRO11(日本向けiPS細胞由来RPE細胞懸濁液又はシート)及びHLCRO12(米国・欧州向けiPS細胞由来RPE細胞懸濁液又はシート)に関しては、医薬品としての承認を取得するために行われる臨床試験(治験)が未だ開始されておりません。そのため人体に対する安全性及び有効性は確認されている訳ではありません。製品の上市(新薬が承認され、市場での販売が開始されること)までには多額の資金と長い年月が必要となり、今後の開発の失敗や遅延のリスクも低くありません。 また、文中の将来に関する事項は、本書提出日現在において当社が判断したものであります。 |
(1)事業の概要
(ア)化合物医薬品分野
①概要
化合物医薬品分野では、国立大学法人九州大学(以下「九州大学」という。)の研究グループが発見したBBG250(Brilliant Blue G-250)という染色性の高い色素を主成分とした眼科手術補助剤を、株式会社産学連携機構九州からの独占的ライセンスに基づき開発しております。
当社は、上記の眼科手術補助剤に関する日本以外の全世界向けの独占的なサブライセンスをDutch Ophthalmic Research Center International B.V.(以下「DORC社」という。)に付与しており、DORC社は、平成22年9月からEU加盟国において、EU加盟国における安全規格に適合しているCEマーキング適合製品として、この眼科手術補助剤を製造・販売しております。この製品は、BBG250の高い染色性を利用して、眼内にある内境界膜又は水晶体を保護するカプセルを一時的に安全に染色し、硝子体・白内障の手術を行いやすくするものです。なお、CEマーキング適合製品とは、欧州における医療機器の製造販売のための必須要求事項を満たしたことを示すCE(フランス語におけるConformite Europeenneの略(英語におけるEuropean Conformity))マークの貼付を許諾された製品のことをいいます。
一方、日本国内については、わかもと製薬株式会社(以下「わかもと製薬」という。)に内境界膜を含む後眼部(網膜等、目の奥に当たる部分)についての独占的サブライセンスを付与しており、わかもと製薬が製造販売承認の取得に向けて開発を進めております。
なお、詳細については、後述「(3)化合物医薬品分野のパイプライン(開発コード:HLM0021、HLM0022、HLM0023)」をご参照ください。
化合物医薬品分野に係る事業系統図は以下のとおりであります。
②収益モデル当社は、上記図に記載のとおり、全世界で特許技術の実施許諾(サブライセンス付与)を行っております。このうち、欧州におけるサブライセンス先であるDORC社は、既にBBG250を使用した眼科手術補助剤を製造・販売しており、当社は、この売上に対してロイヤルティ収入を受け取っています。また、欧州以外の地域においてもサブライセンス先が開発を進めており、そのうち日本以外の地域においてはDORC社から承認を取得した後にロイヤルティ収入等を、日本においてはわかもと製薬からマイルストン収入及び承認を取得した後に製品の販売に応じた収入を得る計画となっています。なお、ロイヤルティ収入とは、契約に基づき、製品が上市された後に、その販売額に応じた一定料率を受領する収入をいい、マイルストン収入とは、契約に基づき、開発の進捗により予め定められた目標(マイルストン)の達成に応じて受領する一時的な収入をいいます。
(イ)iPSC再生医薬品分野
①概要
iPSC再生医薬品は、iPS細胞を分化誘導(細胞を特定の機能を持った細胞、例えば神経細胞・皮膚細胞などに人為的に変化させることをいう。)して作製した健康な細胞を移植することによって、高齢化などにより機能不全に陥った細胞等を置換して機能を回復することを目的としております。
当社では、国立研究開発法人理化学研究所(以下「理研」という。)の髙橋政代プロジェクトリーダー等が中心となって考案したiPS細胞由来の網膜色素上皮細胞(以下「RPE細胞」という。)への分化誘導方法等に関する知見を基にして開発した当社独自のノウハウを用いて、iPS細胞から分化誘導したRPE細胞の効率的な培養方法の確立に成功しました。
そこで、当社は、iPSC再生医薬品として製剤化されたRPE細胞(以下「RPE細胞製品」という。)を使って、根本的な治療法が確立されていない加齢黄斑変性の新たな治療法を開発すべく、より安全かつ効率的な生産方法の確立や治験に向けた準備を進めています。
以下、詳細なパイプラインの説明に先立ち、まず(i)iPS細胞、(ⅱ)RPE細胞と加齢黄斑変性及び(ⅲ)RPE細胞移植による加齢黄斑変性治療法に関してご説明いたします。
(ⅰ)iPS細胞
iPS細胞(人工多能性幹細胞:Induced Pluripotent Stem Cell)とは、平成18年に国立大学法人京都大学(以下「京都大学」という。)の山中伸弥教授が世界で初めて作製に成功し、平成24年にその功績からノーベル生理学・医学賞を受賞したことで広く知られるようになった、皮膚などの体細胞にいくつかの遺伝子(山中因子)を導入することによって作り出される、様々な組織や臓器の細胞に分化する能力(多能性)と、ほぼ無限に増殖する能力(増殖能)を持った細胞であります。
ヒトの体は約60兆個の細胞からなりますが、それらの細胞は全て元々一つの細胞であった受精卵が細胞分裂を繰り返し、それぞれ臓器・器官等を構成する細胞へと分化したものであります。受精卵が特定の細胞に分化していく流れは一方通行であり、従来の技術では一度分化した細胞を分化する前の細胞(幹細胞)に戻すことはできませんでした。ところが、皮膚細胞などの成熟した細胞にいくつかの遺伝子を導入することにより、新たに様々な細胞に分化する能力(多能性)とほぼ無限に増殖する能力(増殖能)を持たせることに成功したものがiPS細胞であります。
当社が開発するiPSC再生医薬品は、このような特徴を有するiPS細胞を特定の細胞に分化誘導し大量培養したうえで、医薬品として製剤化し、人体に移植することで、老化等の原因により機能不全に陥った細胞を健康な細胞に置換し、機能回復を図るものであります。当社は、iPSC再生医薬品の開発においては、当社の強みである①細胞培養、②製薬工業、及び③移植医療という3つの領域の知見が必要となると考えております。
iPSC再生医薬品のように人体の中にある物質を医薬品化した医薬品としては、例えば免疫機能を担っている抗体を用いた抗体医薬品などがありますが、このような医薬品はバイオ医薬品と呼ばれます。当社が開発を進めているiPSC再生医薬品も、生命の最小単位である細胞を用いたバイオ医薬品として、iPS細胞から十分に分化誘導して作製した健康な細胞を移植することによって、臓器移植に近似する治療効果が期待できる点に従来の医薬品と大きく異なる特徴を有しております。
なお、iPS細胞のような多能性幹細胞は、いずれも自然に特定の細胞に分化していく訳ではないため、特定の細胞に分化を誘導するためにはiPS細胞の作製とは別の技術が必要となります。
当社は、この点について、理研の髙橋政代プロジェクトリーダー等が考案したRPE細胞への分化誘導方法及び分化誘導されたRPE細胞等に関する知見を基に、独自の研究開発の結果としてRPE細胞の分化誘導方法及び効率的な培養方法に関するノウハウを蓄積しており、主としてiPS細胞から分化誘導されたRPE細胞をiPSC再生医薬品として提供することによる新しい治療法の実用化についてさらなる研究開発を行っております。
加えて、近年、細胞医薬品分野においては、罹患者自身から採取した細胞(自家細胞)由来の幹細胞を用いたもののみならず、安全性が確認された他人の細胞(他家細胞)由来の幹細胞を活用した医薬品などの研究開発が進んでおります。
当社は、このような流れに沿うものとして、RPE細胞へ分化誘導するために使用するiPS細胞に関して、国内においては、罹患者自身から採取した細胞(自家細胞)から作製されたもの(自家細胞由来iPS細胞)も選択肢には入れつつも、京都大学iPS細胞研究所から提供された他人の細胞(他家細胞)から作製されたもの(他家細胞由来iPS細胞)を用いることを前提として考えております。
なお、iPS細胞と比較される多能性幹細胞としてES細胞(胚性幹細胞:Embryonic Stem Cells。以下「ES細胞」という。)があります。ES細胞は、動物の受精卵から採取した多能性細胞で、同じく多能性と増殖能を備えているものの、受精卵から細胞を採取するという点の倫理的な問題等から国内では過去ヒトに関して研究開発が必ずしも十分に進んでこなかった歴史があります。
(ⅱ) RPE細胞と加齢黄斑変性
網膜は、光や色を感じる視細胞を含む感覚網膜(神経性網膜)と、RPE細胞と呼ばれる組織から構成されます。RPE細胞は、網膜の外側にある一層の細胞で、感覚網膜への栄養補給や老廃物の分解を担っています。そのため、RPE細胞の機能が低下すると視機能を担う感覚網膜の機能も低下してしまいます。
当社が最初の適応症(治療法の対象となる症状をいう。以下同じ。)として治療法の実用化に取り組んでいる加齢黄斑変性(AMD :Age-related Macular Degeneration)は、網膜変性疾患の一種であり、網膜の中でも視力を保つために極めて重要な役割を果たす「黄斑部」に障害が生じる病気で、発症すると次第に視力が低下したり、見え方に異常が生じるなどの症状が現われます。
(黄斑部と網膜色素上皮細胞)
加齢黄斑変性は、滲出型(新生血管型:ウェット型)と非滲出型(萎縮型:ドライ型)に大別され、その原因は、黄斑部を支えるRPE細胞が老化等の原因により感覚網膜への栄養補給や老廃物の分解ができなくなってしまうことにあるものとされております。
日本人に多いウェット型は、黄斑部を支えるRPE細胞の機能不全に伴い、RPE細胞内に貯まった老廃物を分解するために、その外周にある脈絡膜から、脈絡膜新生血管と呼ばれる異常な血管が生えてくるのが特徴であります。この血管は正常な血管とは異なり、もろくて透過性が高いため、破れて出血し、又は水がしみだしてしまうため、網膜が浮腫を起こし、黄斑部の機能が阻害され、視力の低下や視野の歪みなどを生じます。
これに対して、欧米人に多いドライ型は、RPE細胞が加齢により萎縮してしまうことにより、網膜に障害が生じて視力が徐々に低下していく病気であります。
加齢黄斑変性の詳しい発症原因は未だ解明されておらず、根本的な治療法も確立しておりません。加齢黄斑変性は、欧米のような先進国では成人の失明原因として最も多く、公益財団法人難病医学研究財団 難病情報センターのホームページの記載によると、日本での推定罹患者数は平成19年時点で69万人(但し、罹患者数を正確に把握できないため、平成19年に福岡県内の人口約1万人の久山町において行われた調査結果を日本の人口に換算した推定値)と推定されております。
また、米国国立眼病研究所(National Eye Institute)のホームページにおいて公開されている統計データによると、平成22年時点で米国において207万人いると推定される加齢黄斑変性の罹患者は、平成42年(2030年)には366万人に増加すると予測されております。
(ⅲ)RPE細胞移植による加齢黄斑変性治療法
加齢黄斑変性の罹患者に対するRPE細胞の移植治療法は、これまでも多くの移植手術が試されておりましたが、これまでは特に移植対象となる細胞の確保が難しいという点が大きな課題でした。
特に平成18年に英国において罹患者自身の眼から切除して作製したRPE細胞のシートを移植するという治験が行われましたが、一定の有効性が確認されたものの、移植に必要な細胞を眼内の健常な箇所から切除する際の侵襲が大きく一般化はしませんでした。
これに対して、理研の髙橋政代プロジェクトリーダー等は、iPS細胞からRPE細胞を作製するという試みに取り組み、平成25年8月から開始された臨床研究(ヒトを対象とした研究)の中で、平成26年9月に世界で初めて、加齢黄斑変性の罹患者自身の皮膚の細胞から作製したiPS細胞(自家細胞由来iPS細胞)からRPE細胞を分化誘導し、RPE細胞のシートを作製したうえで当該罹患者に移植しました。かかる臨床研究に関して、平成27年3月に行われた日本再生医療学会において、半年経過後もがんはできておらず、細胞シートはきちんと定着しており、問題となる副作用は起きていない、また、異常な血管(上記(ii)の脈絡膜新生血管)を抑える注射治療を行っても続いていた視力低下が移植後には止まっている、と安全性及び有効性を示唆する発表がなされております。
当社のRPE細胞の移植治療法は、このような理研の髙橋政代プロジェクトリーダー等が中心となって考案したiPS細胞からRPE細胞を分化誘導し移植する技術・知見を基礎として、量産化・品質の安定化等に向けた当社独自の技術・知見を加えて開発したものであり、罹患者自身ではない第三者の細胞から作製され、安全性等に関する基準を満たしたiPS細胞(他家細胞由来iPS細胞)から作製したRPE細胞を含む懸濁液(以下「iPS細胞由来RPE細胞懸濁液」という。なお、懸濁液とは液体中に固体粒子が分散しているものをいう。)を注入し、又はiPS細胞由来RPE細胞のシート(以下「iPS細胞由来RPE細胞シート」という。)を移植し、患部に定着させることにより感覚網膜への栄養補給や老廃物の分解機能を回復させ、視機能を改善させる治療法であります。
当社は、日本人に多いウェット型の加齢黄斑変性と欧米人に多いドライ型の加齢黄斑変性の両方を適応症として、この治療法の実用化を目指しております。なお、当社としては、RPE細胞の移植による治療法は、悪くなったRPE細胞を健康なRPE細胞に置換するものであることから、網膜に関する疾患全般に対して効果があるものと期待しております。
以下は、国内におけるiPS細胞の製造からRPE細胞製品の罹患者への投与までの流れを示す図であります。
また、以下は当社が実用化を目指しているiPS細胞由来RPE細胞懸濁液又はシートを用いた加齢黄斑変性の治療法を示す図であります。
② 開発に向けた事業上の取組み
当社は、平成25年2月にiPSアカデミアジャパン株式会社(以下「iPSアカデミアジャパン」という。)との間でRPE細胞を有効成分として含有する細胞製品を対象とする全世界を許諾領域としたiPS細胞樹立基本技術に関する特許実施権許諾契約を締結して非独占的ライセンスを受けるとともに、理研との間で同年3月にiPS細胞を含む多能性幹細胞由来RPE細胞を有効成分として含有する再生医療製品を対象とする全世界を許諾領域とした特許実施許諾契約を締結して独占的ライセンスを受けております。
また、当社は、かかるRPE細胞製品を用いた加齢黄斑変性の治療法の開発を迅速かつ確実に進めるべく、平成25年12月2日、大日本住友製薬株式会社(以下「大日本住友製薬」という。)との間で、日本におけるRPE細胞製品の開発を共同して行うことを合意し、同社との間で①当社の保有する知的財産権の実施許諾に関する実施許諾契約書(サブライセンス契約)、②共同開発を行う上での役割分担や費用負担を定めた共同開発契約書、並びに、③当該製品の製造や販売促進業務を受託する合弁会社の設立と同社への業務委託料等を定めた合弁契約書を締結いたしました。
これらの契約書のうち、実施許諾契約書においては、RPE細胞製品の日本における開発の進捗に伴って当社に支払われるマイルストン収入総額16億円(うち7億円は受領済み)について合意されており、また、共同開発契約書においては、当社がRPE細胞製品の開発に際して必要となる開発費用のうち最大52億円を大日本住友製薬が負担することが合意されております。即ち、当社がRPE細胞製品に関して、日本において平成26年11月25日に施行された医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律(以下「改正薬事法」という。)において再生医療等製品に関して新設された条件及び期限付承認(以下「条件付承認」という。)を取得し、これを販売するためには、本書提出日時点での合理的な見積もりとして平成32年までに、①細胞の特性解析試験・分析法開発・有効性確認試験等を含む前臨床試験や試薬等の消耗品に要する費用、②臨床試験と追加試験に要する費用、③人件費等の販管費などによって構成される開発費用などが必要になる見込みでありますが、当社は、これを当社の自己資金と大日本住友製薬からのマイルストン収入16億円及び同社による最大52億円の開発費用負担によって賄う予定であります。なお、連結損益計算書等における研究開発費の額は、大日本住友製薬による開発費用の負担分を控除した後の金額であります。
当社は、大日本住友製薬との間で、共同開発契約書に基づき、当社がRPE細胞製品の前臨床試験や臨床試験の実施、製造販売承認申請等を行うことに合意しております。他方、RPE細胞製品の製造や販売促進業務に関しては、大日本住友製薬が過去から培ってきた医薬品製造ノウハウや医薬品の販売網等を活かす形が望ましいと判断し、大日本住友製薬との合弁契約書に基づき、両社共同出資により平成26年2月28日付で設立された株式会社サイレジェン(以下「サイレジェン」という。)に対して、国内における製造及び販売促進業務を独占的に委託することに合意しております。
当社は、サイレジェンに対して、当社が考案した効率的な分化誘導方法の技術移管を進めており、承認取得後には、「再生医療等製品の製造管理及び品質管理の基準に関する省令」(以下「GCTP省令」という。なお、GCTPはGood Gene, Cellular, and Tissue-based Products Practiceの略)に従って、京都大学iPS細胞研究所から提供を受けたiPS細胞の親株からRPE細胞製品を製造する業務を委託することを予定しております。
当社は、このようにしてサイレジェンにおいて製造されたRPE細胞製品の独占的な供給を受けて、これを医療機関に販売することを予定しております。
なお、専ら開発に専念し製造・販売をライセンス先の製薬企業に任せるバイオベンチャーの一般的なビジネスモデルとは異なり、当社は、iPS細胞を用いた再生医療を新たな産業として捉え、単に研究開発に留まることなく、当社、サイレジェン及び提携企業によって、製造・販売に至るまでのバリューチェーンの全体を網羅できる体制の構築を目指しており、上述のとおり大日本住友製薬との間でサイレジェンを設立するのみならず、現在は技術者の手作業で行われている細胞培養の負担を軽減し、品質管理や効率化による原価低減の観点から重要と考えられる自動培養装置を、国立大学法人大阪大学(以下「大阪大学」という。)、株式会社ニコン(以下「ニコン」という。)及び澁谷工業株式会社(以下「澁谷工業」という。)との間で共同して開発しております。
さらに、当社は、RPE細胞製品の次のiPSC再生医薬品の候補を探索すべく、再生医療分野において実績を上げる大学・研究機関との共同研究を積極的に進めており、iPS細胞からヒト臓器を作る研究において実績を有する公立大学法人横浜市立大学(以下「横浜市立大学」という。)との間で、iPS細胞等を用いたヒト臓器の再生に関する基礎研究を既に開始しております。かかる基礎研究の内容については、「第2 事業の状況 3 対処すべき課題 (3)製品パイプラインの拡充について」をご参照ください。当社は、将来的に肝臓、腎臓及び膵臓といった臓器の再生も含め、アンメットメディカルニーズ(まだ有効な治療方法が存在していない疾患に対する治療方法開発の必要性)の高い領域についてパイプラインの拡充を模索してまいります。
iPSC再生医薬品分野に係る国内における事業系統図は以下のとおりであります。
(注)平成26年2月に大日本住友製薬との合弁により設立したサイレジェンは、平成26年12月期においては持分法適用関連会社でしたが、平成27年12月期においては連結財務諸表の作成を予定していないため、本書提出日現在においては、持分法適用関連会社ではありません。また、サイレジェンに対するRPE細胞に関するサブライセンス付与は、当社、大日本住友製薬及びサイレジェン間の共同実施許諾契約に基づき、当社及び大日本住友製薬から共同して行われております。
③ 収益モデル
当社は、国内では、共同開発先である大日本住友製薬からマイルストン収入を得るとともに同社による開発費用の負担を得ます。また、当社は、大日本住友製薬との合弁会社であるサイレジェンに製造及び販売促進業務を委託し、サイレジェンに対してこれらの委託費用を支払う一方で、サイレジェンの当社に対する製品売上に対してロイヤルティ収入を得るとともに、サイレジェンから供給を受けたRPE細胞を医療機関に販売することにより製品の販売収入を得る計画です。
収益モデルの概要は以下の図のとおりです。製品開発のリスクに関しましては、「第2 事業の状況 4 事業等のリスク」をご参照ください。また、契約の内容に関しましては「第2 事業の状況 5 経営上の重要な契約等」をご参照ください。
なお、当社が大日本住友製薬から得る予定のマイルストン収入は合計16億円(うち7億円は受領済み)であり、今後、国内における当該製品に関する治験計画届の提出から再生医療等製品に関して新設された条件及び期限付承認(以下「条件付承認」という。)の取得までの複数の目標(マイルストン)の達成により順次受領していく予定です。また、当社は、サイレジェンへの製造委託に基づき、当該製品の薬価の一定割合を製造単価として、サイレジェンから製品の供給を受けます。他方で、当社は、サイレジェンの当社への製品供給に関して、その正味売上高の額の一定割合(大日本住友製薬と同率)をロイヤルティ収入として受領します。さらに、当社は、販売促進業務をサイレジェンに委託し、当該製品の開発費用の総額と前年の年間総売上高の額に応じて変動する販売促進業務に係る業務委託料をサイレジェンに支払います。サイレジェンは、当社と大日本住友製薬が50%ずつの共同出資により設立した合弁会社であることから、サイレジェンの資本及び損益の50%分が実質的に当社に帰属いたします。
当社は、日本において条件付承認を取得した後に条件や期限を付されない承認(以下「本承認」という。)を取得し、また、欧米において製造販売承認を取得するための研究開発資金に充てるため、追加の資金調達を行うことを予定しております。当社は、増資等の方法により欧米における前臨床試験及び臨床試験を自社で継続し、RPE細胞製品の製造販売承認が得られた後に、医療機関への販売による収入を得るか、又は、欧米におけるRPE細胞製品の開発について共同開発先を探した上で米国における当該製品の開発に関する実施許諾契約(ライセンス契約)を締結し、契約締結及びマイルストン達成時のマイルストン収入、並びに上市後のロイヤルティ収入を得るか、のいずれかの方法により収入を得る計画です。
④ 当社の事業の特徴
以上のとおり、当社の事業は、とりわけ以下の点において特徴を有しております。
まず第一に、当社は、RPE細胞へ分化誘導するために使用するiPS細胞に関して、罹患者自身から採取した細胞(自家細胞)から作製されたもの(自家細胞由来iPS細胞)だけではなく、他人の細胞(他家細胞)から作製されたもの(他家細胞由来iPS細胞)を用いることを検討しております。国内においては、京都大学iPS細胞研究所より提供された他家細胞を用いる予定であります。
一般に自家細胞を使用する場合、罹患者が未知のウィルスに感染したり、免疫拒絶反応が起こるリスクは低いものと考えられます。しかしその一方で、罹患者ごとにiPS細胞の作製からRPE細胞の培養までを行う必要があるため、細胞調整に手間がかかり、罹患者個人間のばらつきが大きくなるほか、製造コストが膨らみ製造期間が長くなるなどの問題で多くの罹患者の方々を治療することが困難になると予想されます。さらに、眼内はそもそも免疫拒絶反応が起こりにくい免疫寛容の状態にあるとも言われており、他家細胞を使用する場合であっても、免疫拒絶反応が起こるリスクは高くないと考えられます。また、仮に免疫拒絶反応が起こるとした場合であっても免疫抑制剤を局所的に短期間使用することで対応することも可能と考えられます。
そこで、当社としては、免疫拒絶反応その他の安全面での課題に取り組みつつ、他家細胞から作製されたiPS細胞を用いたRPE細胞の移植による治療方法を確立させることを目指しております。
第二に、治療法実用化後の量産を可能とするため、当社は前述のとおり、iPS細胞の培養及びRPE細胞への分化誘導を担う自動培養装置を、大阪大学、ニコン及び澁谷工業との間で共同して開発しております。この自動培養装置により、現在は技術者の手作業で行われている細胞培養等の負担を軽減し、品質管理・効率化による原価低減を達成することで、上市後の当社製品の幅広い普及を図ることを検討しております。
第三に、当社は、製品の製造供給権を自ら保有し、自社で国内における製造販売承認の取得を目指していることが挙げられます。前述の通り、バイオベンチャーの一般的なビジネスモデルにおいては、自社は専ら開発に専念し、製造・販売をライセンス先の製薬企業に任せ、売上高に応じたロイヤルティ収入を得る例が多くみられます。しかしながら、この収益モデルの場合には一般的に製造・販売を行った製薬企業が利益の大半を獲得する場合が多いと考えられます。
これに対し、当社は、当社、サイレジェン及び提携企業によって、開発から承認取得、製造販売に至るまでのバリューチェーンの全体を網羅する体制を構築することを計画しております。当社は、かかる収益モデルによって、サイレジェンからのロイヤルティ収入に加え、製品の販売に伴う収入も得られる予定です。
(2)当社のパイプライン(製品開発群)
以下の表は、当社の開発品並びにその適応症、市場、開発段階及び本書提出日現在の進捗状況を示しております。
なお、製品の開発に際しては様々なリスクを伴います。当社製品の開発リスクの概要については、「第2 事業の状況 4 事業等のリスク」のとおりであります。
| iPSC再生医薬品分野における主要パイプラインであるHLCRO11及びHLCRO12に関しては、医薬品としての承認を取得するために行われる臨床試験(治験)が未だ開始されておりません。そのため人体に対する安全性及び有効性は確認されている訳ではありません。製品の上市(新薬が承認され、市場での販売が開始されること)までには多額の資金と長い年月が必要となり、今後の開発の失敗や遅延のリスクも低くありません。 |
(注)1.「適応症」のうち、WetAMDとはウェット型の加齢黄斑変性をいい、DryAMDとはドライ型の加齢黄斑変性をいいます。2.「前臨床試験」、「第Ⅰ相試験」、「第Ⅱ相試験」及び「第Ⅲ相試験」とは、改正薬事法に基づき、医薬品の製造販売承認を得るために必要となる試験の各段階を示すものであります(日本におけるiPS細胞由来RPE細胞移植による加齢黄斑変性治療法(HLCR011)は除きます。HLCR011に関しては、注4.をご参照ください。)。前臨床試験とは、臨床試験の前段階として行われる試験であります。第Ⅰ相試験とは、治験薬を少人数の健常者に投与して主として副作用などの安全性を検査する臨床試験です。第Ⅱ相試験とは、少数の罹患者を対象に治験薬の有効性と安全性を検査する臨床試験であり、基礎データを収集し、適応症や使用量等を決定するためのものです。第Ⅲ相試験とは、第Ⅱ相試験の結果を基に、より多くの罹患者を対象に治験薬の有効性と安全性を検査する臨床試験であり、より詳細なデータ収集を治験薬の使用条件等に反映するためのものです。第Ⅰ相試験は、健常者を対象とするものですが、侵襲性が高いなどの理由から最初から罹患者に対して行うべきと考えられる臨床試験に関しては、少人数の罹患者を対象に「第Ⅰ相/第Ⅱ相試験」として、第Ⅰ相試験と第Ⅱ相試験を同一の試験で実施する場合があります。なお、臨床試験とは、ヒトを対象として行われる研究(以下「臨床研究」という。)の中でも特に治療法等のヒトへの有効性・安全性を評価することを目的とするものをいい、特に新薬開発等を目的とした臨床試験を「治験」といいます。
3.「HLM0022」は、サブライセンス先のDORC社が製造販売を行っております。
4.「HLCR011」は、改正薬事法に基づき再生医療等製品に関して新設された条件付承認に関する規定に基づき条件付承認の取得を目指しております。従って、従来の医薬品のような開発の相(第Ⅰ相、第Ⅱ相、第Ⅲ相)の考え方は適用されません。条件付承認とは、再生医療等製品に関して、安全性が確認され、有効性が推定された場合に条件及び期限を付した上で製造販売承認を与える制度で、従来の医薬品のように有効性及び安全性の確認が必要な製造販売承認よりも早い時期に承認の取得が可能になると期待されている制度であります。条件付承認を得た場合には、再生医療等製品の適正な使用の確保のために必要な条件を遵守した上で期限まで製造販売を行うことができます。また、当該再生医療等製品の使用成績に関する資料等を収集した上で、当該期限内に再度製造販売承認の申請を行う必要があり、これにより有効性及び安全性が確認できた場合に改めて本承認が得られることになります。
5.「HLCR012」(米国向け)は、米国における製造販売承認を得るにあたって準拠すべき基準「cGMP」(Current Good Manufacturing Practiceの略)に則った製造のための技術移管に着手しております。
(3)化合物医薬品分野のパイプライン(開発コード:HLM0021、HLM0022、HLM0023)
HLM0021、HLM0022及びHLM0023は、内境界膜又は水晶体を保護するカプセルを一時的に安全に染色し、硝子体・白内障の手術を行いやすくする、BBG250(Brilliant Blue G-250)を主成分とする眼科手術補助剤であります。HLM0021、HLM0022及びHLM0023は、九州大学の研究グループが発見した染色性の高い色素BBG250を基に、当社の代表取締役社長である鍵本忠尚により平成17年に設立されたアキュメンバイオファーマ株式会社(現 アキュメン株式会社。以下「アキュメン」という。)が開発した眼科手術補助剤であり、当社は、平成25年12月20日付でアキュメンからBBG250を含有する眼科手術補助剤に関する事業を譲り受けております。当社は、かかる眼科手術補助剤に関する日本以外の全世界向けの独占的サブライセンスをDORC社に対して付与しており、これによりロイヤルティ収入を受け取っております。また、日本向けの内境界膜染色に関する開発、使用、販売について独占的サブライセンスをわかもと製薬に対して付与しており、これによりマイルストン収入及び製品の販売に応じた収入を得る計画となっております。
眼内は、硝子体というゼリー状の物質で満たされており、その奥に網膜がありますが、網膜剥離等の手術を行うためには、その前段階として、硝子体を切除し、網膜の最表層部分にあり網膜を固く伸ばす機能を有する内境界膜を剥離しなければなりません(このような硝子体の切除を伴う一連の手術を一般的に硝子体手術といいます。)。ところが、内境界膜は、非常に薄く透明な膜であるため、従来は手術経験が豊富な医師以外には剥離が難しいものでした。そこで、当社は、この内境界膜を一時的に青色に染色し、硝子体手術をより安全に行うことを可能にする染色剤としてBBG250を含有する眼科手術補助剤の開発を進めております。
また、白内障は、本来透明であるレンズが老化等の原因で白く濁り硬化することで、視力が低下する病気です。ヒトのレンズは柔らかく、透明な薄い膜でできたカプセルに入っているため、現行の白内障手術ではこの透明なカプセルの真ん中を丸く切り抜き、濁ったレンズの中身を人工の透明なレンズと入れ替えます。そこで、当社は、白内障に関しても、この透明なカプセルを一時的に青色に染色し、手術をより安全に行うことを可能にする染色剤として、同じく眼科手術補助剤の開発を進めております。
HLM0021、HLM0022及びHLM0023の開発方針等は、以下のとおりです。
① HLM0021(日本向け眼科手術補助剤(硝子体手術))
HLM0021(硝子体手術)は、現在日本での第Ⅲ相試験が終了し、製造販売承認の申請に向けて、原薬及び製剤の製造体制の確立、並びに、かかる原薬及び製剤での適切な安定性試験を実施する必要がある眼科手術補助剤(医薬品候補)であり、当社から日本における眼科手術用途(内境界膜染色に限る。)について独占的なサブライセンスを受けた、わかもと製薬により開発が行われております。当社は、治験用の原薬・製剤の供給や必要な試験の実施、製造委託先との調整等の業務を行い、わかもと製薬による内境界膜染色に関するHLM0021の製造販売承認の取得を支援してまいります。
HLM0021は、HLM0022(欧州向け眼科手術補助剤)のようなCEマーキング適合製品という区分ではなく、医薬品と区分されます。
日本において医薬品が製造販売承認を得るためには、(i)「治験薬の製造管理、品質管理等に関する基準」(以下「治験薬GMP」という。なお、GMPはGood Manufacturing Practiceの略)に基づく、ヒトに投与可能な治験薬の準備、(ii)「医薬品の臨床試験の実施の基準に関する省令」(以下「GCP省令」という。なお、GCPはGood Clinical Practiceの略)に基づく、新薬のヒトにおける有効性と安全性を確認するための臨床試験、並びに、(iii)「医薬品及び医薬部外品の製造管理及び品質管理の基準に関する省令」(以下「医薬品GMP」という。)に基づく、製造販売承認を得た後に医薬品の製造及び品質を適切に管理できる体制の具備といった要件を充足する必要があります。
もっとも、HLM0021の主成分は、平成22年に欧州のCEマーキング適合製品として有効性・安全性の確認を得て製造販売が行われているHLM0022の主成分と同じ化合物BBG250であります。そこで、(i)国内でHLM0022の製剤を用いて第Ⅲ相試験を実施すること、(ii)医薬品GMPに基づく原薬及び製剤を製造する体制を確立し、原薬及び製剤について安定性試験を実施すること、並びに、(iii)第Ⅲ相試験で得られた臨床試験の成績と今まで欧州で用いられた安全性に関する成績を比較した結果、医薬品GMPに基づき製造された製剤の品質がHLM0022の品質と同様であることについての適切な説明を申請資料中に記載することの3つの条件を満たすことによって製造販売承認の申請が行われる予定です。
このうち、HLM0022の製剤を用いた第Ⅲ相試験については、既に九州大学が中心となった医師主導治験が実施され、平成26年10月には、硝子体手術時の内境界膜の可視化に有効であり、手術の容易性が向上することが確認されるとともに、臨床的に重要な安全性の問題は認められなかった、との結果が得られております。また、当社は、医薬品GMPに基づく新たな原薬の合成法の技術開発を終え、原薬製造体制の確立に向けての検討を行っており、かかる製造体制が確立された後、原薬の安定性試験を実施する予定です。一方、製剤の開発に関しましては、前述の原薬を用いて、CEマーキング適合製品(HLM0022)を製造している受託製造会社において製剤の作製を行うにあたっての課題の抽出が終了し、製剤の製造に向けての準備が行われております。製剤に関しても製造体制が確立された後、原薬と同様に安定性試験が実施される予定であります。
以上から、今後、医薬品GMPに基づく原薬及び製剤の製造体制が確立され、原薬及び製剤について1年間分の安定性試験のデータが得られた時点で、安全性に関する成績の比較等を行うことで、製造販売承認の申請が可能になるものと考えております。
以上を前提に、当社としては、当社委託先による医薬品GMPに合致した原薬及び製剤の1年間分の安定性試験のデータが得られると見込まれる平成29年第1四半期頃に、製造販売承認申請が可能になると見込んでおります。但し、「第2 事業の状況 4 事業等のリスク」に記載のとおり、製品開発に際しては様々なリスクが伴うため、当社として上記の各予定時期を保証できるものではありません。また、実際の開発はわかもと製薬の判断によって行われることになります。このため、当社として現段階で製造販売承認申請の具体的な時期又はわかもと製薬による最終的な製造販売承認の取得を保証することはできません。
② HLM0021(日本向け眼科手術補助剤(白内障手術))
HLM0021(白内障手術)は、現在、当社が日本国内において白内障手術向けのサブライセンス先の選定を行っている眼科手術補助剤(医薬品候補)であります。当社は、かかる適応症に関しても今後サブライセンス先による治験、製造販売承認の取得を目指す予定であります。但し、現時点で具体的なサブライセンス先は決定しておらず、また、実際の開発はサブライセンス先の判断によって行われることになります。加えて、「第2 事業の状況 4 事業等のリスク」に記載のとおり、製品開発に際しては様々なリスクが伴います。このため、当社として現段階で今後の開発スケジュールを明言し、又はサブライセンス先による最終的な製造販売承認の取得を保証することはできません。
③ HLM0022(欧州向け眼科手術補助剤:商品名「ILM-Blue」)
HLM0022は、BBG250に関する日本を除く全世界向けの独占的なサブライセンスを付与しているDORC社により、欧州の必須安全要求事項(ESRs:Essential Safety Requirements)に適合したことを示すCEマーキング適合製品としてEU加盟国において平成22年9月から製造販売が行われている、硝子体手術向けの眼科手術補助剤であります。
④ HLM0023(米国向け眼科手術補助剤)
HLM0023は、BBG250に関する日本を除く全世界向けの独占的なサブライセンスを付与しているDORC社により開発が進められている米国向けの眼科手術補助剤(医薬品候補)であります。
HLM0023は、日本向けのHLM0021と同様に米国において医薬品と区分されますが、同国における医薬品の製造販売承認申請にあたっては、一般に、日本の医薬品GMPに対応する米国の基準(cGMP)に則った製剤を用いて治験を実施することが求められます。
開発を行うDORC社からは、米国の食品医薬品局(以下「FDA」という。なお、FDAはFood and Drug Administrationの略)に対する相談の結果、欧州におけるHLM0022の臨床成績を使用することが認められ、現在、第Ⅲ相試験に関する検討作業を行っていると聞いております。
もっとも、米国における第Ⅲ相試験の具体的な開始時期等及び製造販売承認の申請時期は、現在検討中の製造法が確定した時点で、実際の開発を行うDORC社によって最終的に決定されます。また、「第2 事業の状況 4 事業等のリスク」に記載のとおり、製品開発に際しては様々なリスクが伴います。このため、当社として現段階でこれらの具体的な予定時期を明言し、又はDORC社による最終的な製造販売承認の取得を保証することはできません。
(4)iPSC再生医薬品分野のパイプライン(開発コード:HLCR011、HLCR012)
HLCR011(日本向け)及びHLCR012(欧米向け)は、平成25年6月から日本において前臨床試験を開始し、治験に向けた準備を行っている再生医療等製品の候補であります。
当社は、平成25年2月にiPSアカデミアジャパンとの間でRPE細胞を有効成分として含有する細胞製品を対象とする全世界を許諾領域としたiPS細胞樹立基本技術に関する特許実施権許諾契約(非独占)を締結するとともに、同年3月に理研との間でiPS細胞を含む多能性幹細胞由来RPE細胞を有効成分として含有する再生医療製品を対象とする全世界を許諾領域とした特許実施許諾契約(独占)を締結しております。
当社は、HLCR011(日本向け)に関して、大日本住友製薬と平成25年12月2日付で実施許諾契約書、共同開発契約書及び合弁契約書を締結しており、共同開発先である大日本住友製薬から開発の進捗に応じてマイルストン収入合計16億円(うち7億円は受領済み)を得るほか、最大52億円までの開発費用の負担を得ることに合意しております。製造販売承認の取得後には、製剤化されたRPE細胞を医療機関に販売することにより医薬品販売収入を得るとともに、大日本住友製薬との共同出資による合弁会社であるサイレジェンに製造及び販売促進業務を委託し、同社から製品の当社への供給に関してロイヤルティ収入を得る計画です。
なお、当社は、HLCR011(日本向け)について条件付承認を取得した後に本承認を取得し、また、HLCR012(米国向け)及びHLCR012(欧州向け)の製造販売承認を取得するための研究開発資金に充てるため、追加の資金調達を行うことを予定しております。当社は、増資等の方法により欧米における前臨床試験及び臨床試験を自社で継続し、RPE細胞製品の製造販売承認が得られた後に、医療機関への販売による収入を得るか、又は、欧米におけるRPE細胞製品の開発について共同開発先と実施許諾契約(ライセンス契約)を締結しマイルストン収入及び上市後のロイヤルティ収入を得るか、のいずれかの方法により収入を得る計画です。
① HLCR011(日本向けiPS細胞由来RPE細胞懸濁液又はシート)
HLCR011は、iPS細胞を正常なRPE細胞に分化誘導し、純化した上で、iPS細胞由来RPE細胞懸濁液又はiPS細胞由来RPE細胞シートという形で罹患者に移植し、加齢黄斑変性の治療を行うiPSC再生医薬品候補であります。
iPS細胞由来RPE細胞の懸濁液の注射による治療法については、平成26年10月にES細胞由来RPE細胞を用いたAdvanced Cell Technology, Inc.(現 Ocata Therapeutics, Inc.)が実施した米国での臨床成績が発表されており、第Ⅰ相/第Ⅱ相試験にて良好な安全性が示され、有効性が推定されています。そこで、当社においてもiPS細胞由来RPE細胞懸濁液による治療法の開発を優先的に検討しております。
なお、当社のiPS細胞由来RPE細胞懸濁液による治療法は、理研の髙橋政代プロジェクトリーダー等が中心となって考案したiPS細胞からRPE細胞を分化誘導し移植する技術・知見を基礎として、量産化・品質の安定化等に向けた当社独自の技術・知見を加えて開発したものであり、細胞培養・分化誘導等の技術は基本的に同じであるものの、①罹患者自身ではない第三者の細胞から作製され、安全性等に関する基準を満たしたiPS細胞(他家細胞由来iPS細胞)を使用する点、②RPE細胞のシートではなく、RPE細胞を含む懸濁液を優先的に検討している点、③量産化及び条件付承認の取得のための基準への適合を目的とした工程変更が加えられている点等において異なるものであります。また、当社は、理研の髙橋政代プロジェクトリーダー等の臨床研究とは別個に改正薬事法に基づき厳格な基準に則った臨床試験(治験)を新たに実施しなければなりません。
HLCR011は、改正薬事法において再生医療等製品と区分されるものです。再生医療等製品は、化合物医薬品と異なり、数多くの細胞を培養した結果として作製された均質でない細胞を用いた医薬品であることから、全ての治験薬の成分が同質であることを前提とした従来の医薬品のための治験手続を経ることを求めた場合には、より多くの人数を対象とした臨床試験で安全性と有効性の確認が必要となり、開発の難易度が高まるとともに開発期間も長期に亘る上、これに伴うコスト増の結果、開発そのものに対するハードルが大幅に高くなります。そこで、かかる問題を是正するため、改正薬事法は、新たに再生医療等製品の性質により適した制度として条件付承認制度を設けており、この結果、再生医療等製品の製造販売承認の申請者は、(i)安全性・有効性を確認するに足りる十分な症例数の臨床試験を通じて行われる、従来の医薬品に準じた承認申請手続によって、条件や期限を付されない承認(以下「本承認」という。)を受けるプロセスのほか、(ii)より少人数を対象とした臨床試験を通じて、安全性の確認と有効性の推定を得ることにより、条件及び期限を付された条件付承認を受けるプロセスを活用することができるようになっております。
かかる条件付承認制度は、条件及び期限の設定により罹患者の安全に配慮しつつ、罹患者の新薬へのアクセスを早期に実現するものであり、かつ、申請者において罹患者に対する再生医療等製品の使用成績等の情報を蓄積し、安全性及び有効性を迅速に確認することを可能とするものであります。少ない症例数の臨床試験の試験成績から安全性の確認及び有効性の推定を得て条件付承認を受けた申請者は、かかる条件を遵守した上で期限まで再生医療等製品を実際に罹患者に使用し、以後その使用の成績等に関する調査を行った上で、期限内に改めて製造販売承認の申請をしなければならず、その結果、安全性の確認及び有効性の確認が得られれば条件及び期限の付されない本承認を受けることができます。
当社は、上記のような条件付承認制度を活用することを念頭に、再生医療等製品の候補としてのHLCR011について平成29年から第Ⅰ相/第Ⅱ相試験に相当する臨床試験を数十症例程度の規模で開始する予定であり、そのうち一部の罹患者に対する投与が完了し、投与後数か月分の試験成績の蓄積があった時点、即ち、最も早い場合で平成31年下期頃に製造販売に係る条件付承認の申請を行う予定です。そして、審査期間中に、残りの罹患者への投与後数か月分の試験成績を追加提出する予定です。但し、必要となる症例数、データ蓄積期間、及びこのような試験成績の追加提出を前提とする条件付承認の申請の可否については、現在何ら指針がなく、具体的なスケジュールと併せて、第Ⅰ相/第Ⅱ相試験の実施に先立ち、平成28年中を目途として医薬品や医療機器などの製造販売承認審査及び治験等に関する指導・助言を行う独立行政法人医薬品医療機器総合機構(以下「PMDA」という。なお、PMDAとはPharmaceutical and Medical Devices Agencyの略)と対面助言を行った上で確定される予定であります。
さらに、当社は、条件付承認を受けることができた場合、新たに市販後臨床試験を実施するのではなく、HLCR011を投与した罹患者に対する市販後調査を行い、その成績をもって本承認の取得に向けた製造販売承認の再申請を行うことができるか検討中ですが、これについては、平成28年中を目途とした上記のPMDAとの対面助言の中で併せて協議を行い、最終的には承認申請を行った後の承認審査の過程で確定される予定であります。
以上のとおり、当社は、既存の治験実務に従って目標とすべき各段階の予定時期を設定しておりますが、改正薬事法に基づく条件付承認制度を活用した治験手続に関しては未だ運用実績がないことから明らかでない点も多く、また、「第2 事業の状況 4 事業等のリスク」に記載のとおり、製品開発に際しては様々なリスクが伴うため、当社として上記各予定時期又は当該製品に関する製造販売承認の取得を保証できるものではありません。
なお、かかる治療法は、現在のところ前臨床試験を実施している段階にあり、未だ臨床試験も開始されておらず、ヒトに対する有効性及び安全性が確認されておりません。さらに条件付承認の条件が解除されるまでに現時点から10年以上の期間を要する可能性もあり、今後開発の失敗や遅延のリスクも低くはありません。
② HLCR012(米国・欧州向けiPS細胞由来RPE細胞懸濁液又はシート)
HLCR012は、ドライ型加齢黄斑変性を適応症としたiPS細胞由来RPE細胞懸濁液又はシートの移植による治療法であり、米国・欧州におけるiPSC再生医薬品候補であります。
米国・欧州においては、日本において平成26年11月25日に施行された改正薬事法に基づく条件付承認制度と同様の制度が存在しないため、従来の医薬品同様、第Ⅰ相試験から第Ⅲ相試験までの臨床試験を経て、各国の薬事法に基づく製造販売承認申請を行う必要があります。
当社は、調達資金を研究開発費に充当することによって、米国におけるRPE細胞製品に係る前臨床試験を継続する計画です。
このため、当社は、まずは米国において、医薬品受託製造会社(以下「CMO」という。)に対して、治験及び承認後の販売に使用されるiPS細胞由来RPE細胞の培養法及び品質管理試験方法についての技術移管を行っているところであります。
当社は、HLCR011(日本向け)について条件付承認を取得した後に本承認を取得し、また、HLCR012(米国向け)及びHLCR012(欧州向け)の製造販売承認を取得するための研究開発資金に充てるため、追加の資金調達を行うことを予定しております。なお、当社は、追加の資金調達の方法として、増資のほか、欧米におけるRPE細胞製品の開発について共同開発先を探したうえで実施許諾契約(ライセンス契約)を締結し、契約締結及びマイルストン達成時のマイルストン収入、並びに上市後のロイヤルティ収入を得ることなども柔軟に検討してまいります。
HLCR012(米国向け)の具体的なスケジュールについては、今後、臨床試験実施のための米国FDAとの事前会議等を通じて検討していく予定です。
また、欧州については、米国の第Ⅰ相/第Ⅱ相試験の結果を使って、第Ⅲ相試験から試験を実施することを検討しております。
なお、「第2 事業の状況 4 事業等のリスク」に記載のとおり、製品開発に際しては様々なリスクが伴うため、当社として上記各予定時期又は当該製品に関する製造販売承認の取得を保証できるものではありません。また、かかる治療法は、現在のところ前臨床試験を実施している段階にあり、未だ臨床試験も開始されておらず、ヒトに対する有効性及び安全性が確認されておりません。さらに欧米において承認を取得するまでに現時点から10年以上の期間を要する可能性もあり、今後開発の失敗や遅延のリスクも低くはありません。